![]() Проведен количественный анализ экспериментальных катодных поляризационных кривых, полученных растворах 0,33 мМ К3[Fe(CN)6] + 0 – 1,5 мМ КСl на ртутном электроде. C использованием результатов квантово-химического расчета геометрии и зарядовых распределений в частицах [Fe(CN)6]3– и [Fe(CN)6]4–, показано, что взаимодействие этих частиц с полем двойного электрического слоя эквивалентно отталкиванию эффективных точечных зарядов, расположенных вблизи центров анионов в диффузном. Рассчитаны эффективный борновский радиус феррицианид-аниона и энергия реорганизации растворителя, а также даны оценки внутрисферной составляющей энергии реорганизации. С использованием этих модельных параметров по уравнению квантово-механической теории элементарного акта рассчитаны теоретические зависимости коэффициента переноса a от перенапряжения. Показано, что определяемая из исправленных тафелевских зависимостей величина a существенно зависит от предположений, принятых при анализе системы в рамках классической теории замедленного разряда и близка к теоретической только при учете участия в реакции анион-катионных ассоциатов, образующихся в объеме раствора. Одновременно в рамках такого подхода находит объяснение и слабая зависимость скорости процесса от температуры. Таким образом, экспериментальные факты не противоречат теоретическому прогнозу о протекании реакции в окрестности безактивационной области.
Проведен количественный анализ экспериментальных катодных поляризационных кривых, полученных растворах 0,33 мМ К3[Fe(CN)6] + 0 – 1,5 мМ КСl на ртутном электроде. C использованием результатов квантово-химического расчета геометрии и зарядовых распределений в частицах [Fe(CN)6]3– и [Fe(CN)6]4–, показано, что взаимодействие этих частиц с полем двойного электрического слоя эквивалентно отталкиванию эффективных точечных зарядов, расположенных вблизи центров анионов в диффузном. Рассчитаны эффективный борновский радиус феррицианид-аниона и энергия реорганизации растворителя, а также даны оценки внутрисферной составляющей энергии реорганизации. С использованием этих модельных параметров по уравнению квантово-механической теории элементарного акта рассчитаны теоретические зависимости коэффициента переноса a от перенапряжения. Показано, что определяемая из исправленных тафелевских зависимостей величина a существенно зависит от предположений, принятых при анализе системы в рамках классической теории замедленного разряда и близка к теоретической только при учете участия в реакции анион-катионных ассоциатов, образующихся в объеме раствора. Одновременно в рамках такого подхода находит объяснение и слабая зависимость скорости процесса от температуры. Таким образом, экспериментальные факты не противоречат теоретическому прогнозу о протекании реакции в окрестности безактивационной области.
![]() Редокссистема [Fe(CN)6]3–/[Fe(CN)6]4– — без преувеличения — наиболее детально исследованная модельная система в кинетике гомогенных процессов переноса электрона, а также классический объект электрохимической кинетики. Электродные превращения ферро/феррицианида вблизи равновесного потенциала можно исследовать только на адсорбционно активных благородных металлах. При этом имеют место существенные искажения частиц реагентов и продуктов в результате хемосорбционных взаимодействий (не исключена, в частности, диссоциативная адсорбция) [1–3], что затрудняет сопоставление с гомогенными процессами. При высоких перенапряжениях электровосстановление [Fe(CN)6]3– было впервые изучено А.Н.Фрумкиным и Г.М.Флорианович на ртутном электроде в разбавленных растворах фона [4] (Работа посвящена юбилею профессора Г.М.Флорианович.). Была найдена аномалия поляризационной кривой (спад тока при потенциалах отрицательнее пнз и выход на плато), исчезающая при добавлении индифферентного электролита. Предельный диффузионный ток наблюдался уже при концентрациях фона порядка 1 мМ.
Редокссистема [Fe(CN)6]3–/[Fe(CN)6]4– — без преувеличения — наиболее детально исследованная модельная система в кинетике гомогенных процессов переноса электрона, а также классический объект электрохимической кинетики. Электродные превращения ферро/феррицианида вблизи равновесного потенциала можно исследовать только на адсорбционно активных благородных металлах. При этом имеют место существенные искажения частиц реагентов и продуктов в результате хемосорбционных взаимодействий (не исключена, в частности, диссоциативная адсорбция) [1–3], что затрудняет сопоставление с гомогенными процессами. При высоких перенапряжениях электровосстановление [Fe(CN)6]3– было впервые изучено А.Н.Фрумкиным и Г.М.Флорианович на ртутном электроде в разбавленных растворах фона [4] (Работа посвящена юбилею профессора Г.М.Флорианович.). Была найдена аномалия поляризационной кривой (спад тока при потенциалах отрицательнее пнз и выход на плато), исчезающая при добавлении индифферентного электролита. Предельный диффузионный ток наблюдался уже при концентрациях фона порядка 1 мМ.
![]() В дальнейшем выяснению детального хода кривых при высоких отрицательных зарядах уделялось значительное внимание [4–6], в частности, исследовалось влияние природы катиона и температуры на скорость процесса. Отметим, что количественно описать поляризационную кривую электровосстановления [Fe(CN)6]3– и ход исправленных тафелевских зависимостей (ИТЗ) не удавалось даже в предположении о чрезвычайно низких величинах коэффициента переноса, хотя внешнесферный характер реакции электровосстановления [Fe(CN)6]3– на ртутном и ртутеподобных электродах [5,6] не вызывал сомнений.
В дальнейшем выяснению детального хода кривых при высоких отрицательных зарядах уделялось значительное внимание [4–6], в частности, исследовалось влияние природы катиона и температуры на скорость процесса. Отметим, что количественно описать поляризационную кривую электровосстановления [Fe(CN)6]3– и ход исправленных тафелевских зависимостей (ИТЗ) не удавалось даже в предположении о чрезвычайно низких величинах коэффициента переноса, хотя внешнесферный характер реакции электровосстановления [Fe(CN)6]3– на ртутном и ртутеподобных электродах [5,6] не вызывал сомнений.
![]() Обсуждалась гипотеза о нарушении равновесного строения двойного слоя при высоких перенапряжениях [7,8]. На основании оценок, выполненных недавно в [9], высказано предположение о том, что в области высоких перенапряжений происходит практически безактивационный перенос электрона(Здесь и далее под окрестностью безактивационной области мы подразумеваем интервал перенапряжений, в котором нарушена линейная зависимость a от перенапряжения, подробнее см. в [9].). На пути количественного анализа проблемы встречаются определённые трудности, в первую очередь связанные с необходимостью учесть и обьяснить значительное влияние природы катиона электролита фона [5].
Обсуждалась гипотеза о нарушении равновесного строения двойного слоя при высоких перенапряжениях [7,8]. На основании оценок, выполненных недавно в [9], высказано предположение о том, что в области высоких перенапряжений происходит практически безактивационный перенос электрона(Здесь и далее под окрестностью безактивационной области мы подразумеваем интервал перенапряжений, в котором нарушена линейная зависимость a от перенапряжения, подробнее см. в [9].). На пути количественного анализа проблемы встречаются определённые трудности, в первую очередь связанные с необходимостью учесть и обьяснить значительное влияние природы катиона электролита фона [5].
![]() Влияние природы катиона на скорость процесса, а также низкие, зависящие от природы катиона, энергии активации, формально отвечающие близости к безактивационной области, получили лишь качественное объяснение в рамках представлений о возрастании специфической адсорбции катионов в ряду Li+<Na+<K+<Сs+ и образовании катионных мостиков в двойном электрическом слое. Отметим, что скорость гомогенного процесса [Fe(CN)6]3–+[Fe(CN)6]4– также зависит от природы катиона и растёт при переходе от Li+ к Сs+ [10]. В настоящей работе предпринята попытка рассмотреть классические экспериментальные данные [5,6] с учётом взаимодействий анионного реагента с катионами фонового электролита в объёме раствора и в приэлектродном слое и уточнить на этой основе возможные зависимости коэффициента переноса от перенапряжения. Полученные зависимости сопоставляются с модельными, построенными на основании квантово-механической теории переноса электрона. Анализируются и экспериментальные данные [11] по температурной зависимости скорости процесса.
Влияние природы катиона на скорость процесса, а также низкие, зависящие от природы катиона, энергии активации, формально отвечающие близости к безактивационной области, получили лишь качественное объяснение в рамках представлений о возрастании специфической адсорбции катионов в ряду Li+<Na+<K+<Сs+ и образовании катионных мостиков в двойном электрическом слое. Отметим, что скорость гомогенного процесса [Fe(CN)6]3–+[Fe(CN)6]4– также зависит от природы катиона и растёт при переходе от Li+ к Сs+ [10]. В настоящей работе предпринята попытка рассмотреть классические экспериментальные данные [5,6] с учётом взаимодействий анионного реагента с катионами фонового электролита в объёме раствора и в приэлектродном слое и уточнить на этой основе возможные зависимости коэффициента переноса от перенапряжения. Полученные зависимости сопоставляются с модельными, построенными на основании квантово-механической теории переноса электрона. Анализируются и экспериментальные данные [11] по температурной зависимости скорости процесса.
![]() Квантовохимические расчеты гексацианокомплексов железа предпринимались ранее в работах [12] (метод INDO) и [13] (метод ca-дискретного варьирования). Отметим, однако, что полуэмпирические методы, основанные на идее частичного пренебрежения дифференциальным перекрыванием (INDO), не всегда надежны для описания изменения структуры комплексов при переходе от одной степени окисления центрального атома к другой. Кроме того, используемая обычно для d-металлов параметризация нередко приводит к переоценке обменных взаимодействий и, как следствие, к ошибочному предсказанию мультиплетности основного состояния. С другой стороны, применение одного из самых простых локальных функционалов типа «ca» целесообразно, главным образом, для систем с фиксированной молекулярной геометрией (в работе [13] использовались экспериментальные данные по длинам связей в комплексах [Fe(CN)6]3– и [Fe(CN)6]4–), что также ограничивает круг задач, решаемых методом ca).
Квантовохимические расчеты гексацианокомплексов железа предпринимались ранее в работах [12] (метод INDO) и [13] (метод ca-дискретного варьирования). Отметим, однако, что полуэмпирические методы, основанные на идее частичного пренебрежения дифференциальным перекрыванием (INDO), не всегда надежны для описания изменения структуры комплексов при переходе от одной степени окисления центрального атома к другой. Кроме того, используемая обычно для d-металлов параметризация нередко приводит к переоценке обменных взаимодействий и, как следствие, к ошибочному предсказанию мультиплетности основного состояния. С другой стороны, применение одного из самых простых локальных функционалов типа «ca» целесообразно, главным образом, для систем с фиксированной молекулярной геометрией (в работе [13] использовались экспериментальные данные по длинам связей в комплексах [Fe(CN)6]3– и [Fe(CN)6]4–), что также ограничивает круг задач, решаемых методом ca).
![]() В настоящей работе квантовохимические расчеты комплексов [Fe(CN)6]3–/4–проводились с помощью трех различных методов: Хартри-Фока-Рутана (SCF) в комбинации с неограниченным методом Хартри-Фока для систем с открытой оболочкой; теории возмущений Меллера-Плессета второго порядка (MP2); теории функционала плотности (метод Кона-Шема) с использованием «гибридного» функционала B3LYP [14,15,16]. Для описания 3s3p3d4s4p электронов атома Fe был использован дважды расщепленный (DZ) базис [17]. Остальные электроны (1s2s2p) включались в эффективный остовный потенциал Хея-Вадта [17]. Валентная электронная конфигурация атомов C и N (2s2p) описывалась базисным набором Даннинга-Хузинаги (D95V) [18]. Тестовые расчеты проводились также с базисом 6-31g (d) [19] для атомов азота и углерода. Геометрия комплексов оптимизировалась градиентным методом. Все квантовохимические расчеты проводились с использованием программных пакетов Gaussian 94, 98 [16].
В настоящей работе квантовохимические расчеты комплексов [Fe(CN)6]3–/4–проводились с помощью трех различных методов: Хартри-Фока-Рутана (SCF) в комбинации с неограниченным методом Хартри-Фока для систем с открытой оболочкой; теории возмущений Меллера-Плессета второго порядка (MP2); теории функционала плотности (метод Кона-Шема) с использованием «гибридного» функционала B3LYP [14,15,16]. Для описания 3s3p3d4s4p электронов атома Fe был использован дважды расщепленный (DZ) базис [17]. Остальные электроны (1s2s2p) включались в эффективный остовный потенциал Хея-Вадта [17]. Валентная электронная конфигурация атомов C и N (2s2p) описывалась базисным набором Даннинга-Хузинаги (D95V) [18]. Тестовые расчеты проводились также с базисом 6-31g (d) [19] для атомов азота и углерода. Геометрия комплексов оптимизировалась градиентным методом. Все квантовохимические расчеты проводились с использованием программных пакетов Gaussian 94, 98 [16].
![]() Длины связей в исследуемых комплексах, рассчитанные различными методами, приведены в Tаблице 1. Как видно из таблицы, результаты расчетов комплекса [Fe(CN)6]3– методом SCF предсказывают заметно выраженный эффект Яна-Теллера («сплюснутый» октаэдр). Этот результат находится в противоречии с экспериментальными данными, свидетельствующими об октаэдрической симметрии лигандного окружения [Fe(CN)6]3– [20]. Однако учет электронной корреляции на основе теории функционала плотности делает ян-теллеровское искажение пренебрежимо малым: длины аксиальных и экваториальных связей Fe–C практически совпадают (Таблица 1, рис. 1а). Кроме того, результаты, полученные методом B3LYP, не подтверждают вывод о значительной внутримолекулярной реорганизации комплексов [Fe(CN)6]3–/4–, следующий из расчетов SCF. Таким образом, учет электронной корреляции в изучаемых системах играет важную роль.
Длины связей в исследуемых комплексах, рассчитанные различными методами, приведены в Tаблице 1. Как видно из таблицы, результаты расчетов комплекса [Fe(CN)6]3– методом SCF предсказывают заметно выраженный эффект Яна-Теллера («сплюснутый» октаэдр). Этот результат находится в противоречии с экспериментальными данными, свидетельствующими об октаэдрической симметрии лигандного окружения [Fe(CN)6]3– [20]. Однако учет электронной корреляции на основе теории функционала плотности делает ян-теллеровское искажение пренебрежимо малым: длины аксиальных и экваториальных связей Fe–C практически совпадают (Таблица 1, рис. 1а). Кроме того, результаты, полученные методом B3LYP, не подтверждают вывод о значительной внутримолекулярной реорганизации комплексов [Fe(CN)6]3–/4–, следующий из расчетов SCF. Таким образом, учет электронной корреляции в изучаемых системах играет важную роль.
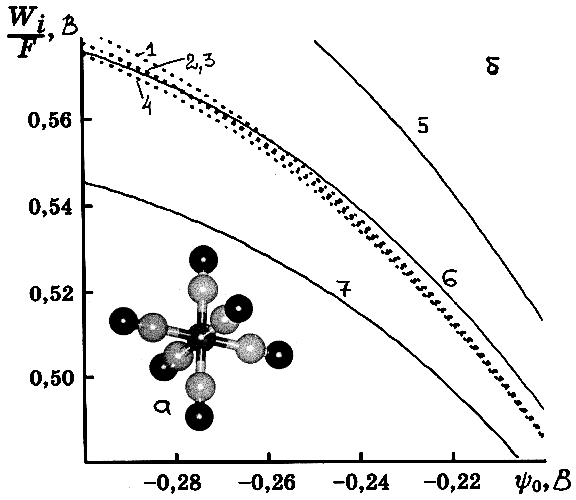 |
| Рис. 1. Строение реагента (а) и зависимости микроскопических работ сближения (Wi/F) от yo, рассчитанные для наборов точечных зарядов, локализованных на атомах реагента (1–4) и на единичных точечных зарядах (z=–3) (5–7) при их смещении от внешней плоскости Гельмгольца на расстоянии x: 5 — 0.3; 6 — 0.4; 7 — 0.5 нм. Кривые 1–4 отвечают разным версиям зарядовых распределений (NPA и по Малликену) и двум возможным ориентациям реагента (одна или две CN-группы «касаются» плоскости Гельмгольца. |
![]() Как видно из Табл. 1, геометрические характеристики [Fe(CN)6]4–, полученные методами B3LYP и MP2, различаются несущественно. Учет внутренних электронов (1s) и поляризационных d-орбиталей для атомов C и N (базис 6-31g(d,р)) также не приводит к заметным изменениям в рассчитываемых величинах.
Как видно из Табл. 1, геометрические характеристики [Fe(CN)6]4–, полученные методами B3LYP и MP2, различаются несущественно. Учет внутренних электронов (1s) и поляризационных d-орбиталей для атомов C и N (базис 6-31g(d,р)) также не приводит к заметным изменениям в рассчитываемых величинах.
![]() Как следует из результатов расчетов, длина связи Fe-С в восстановленной форме гексацианоферрата больше по сравнению с рассчитанной для окисленной формы комплекса, что противоречит экспериментальным данным по структуре цианидных комплексов в кристаллах (r(Fe–C)=0.1913¸0.1926 нм [Fe(CN)6]3– и (r(Fe–C)=0.189¸0.190 нм [Fe(CN)6]4–) [21]. Действительно, перенос электрона на связывающую T2g орбиталь комплекса [Fe(CN)6]3– должен приводить к дополнительной стабилизации молекулы и, следовательно, к укорачиванию длины связи Fe–C. На наш взгляд, в расчетах изолированных гексацианоферратов этот эффект не воспроизводится по следующей причине. Как показывают результаты вычислений, энергия высших занятых молекулярных орбиталей (ВЗМО) анионных комплексов в газовой фазе является положительной вследствие их высокого отрицательного заряда. В то же время учет гидратации в рамках континуальной модели PCM (Point Charge Model) на основе самосогласованной теории реактивного поля (см., например, обзор [22]) с использованием оптимальной геометрии цианидных комплексов железа, рассчитанной для газовой фазы, приводит к значительному понижению энергии ВЗМО (на 10.4 эВ и 13.9 эВ для [Fe(CN)6]3–и [Fe(CN)6]4–, соответственно) и изменению её знака. Несмотря на то, что для кон-шемовских орбиталей теорема Купманса не применима, их энергию допустимо использовать для выводов качественного характера.
Как следует из результатов расчетов, длина связи Fe-С в восстановленной форме гексацианоферрата больше по сравнению с рассчитанной для окисленной формы комплекса, что противоречит экспериментальным данным по структуре цианидных комплексов в кристаллах (r(Fe–C)=0.1913¸0.1926 нм [Fe(CN)6]3– и (r(Fe–C)=0.189¸0.190 нм [Fe(CN)6]4–) [21]. Действительно, перенос электрона на связывающую T2g орбиталь комплекса [Fe(CN)6]3– должен приводить к дополнительной стабилизации молекулы и, следовательно, к укорачиванию длины связи Fe–C. На наш взгляд, в расчетах изолированных гексацианоферратов этот эффект не воспроизводится по следующей причине. Как показывают результаты вычислений, энергия высших занятых молекулярных орбиталей (ВЗМО) анионных комплексов в газовой фазе является положительной вследствие их высокого отрицательного заряда. В то же время учет гидратации в рамках континуальной модели PCM (Point Charge Model) на основе самосогласованной теории реактивного поля (см., например, обзор [22]) с использованием оптимальной геометрии цианидных комплексов железа, рассчитанной для газовой фазы, приводит к значительному понижению энергии ВЗМО (на 10.4 эВ и 13.9 эВ для [Fe(CN)6]3–и [Fe(CN)6]4–, соответственно) и изменению её знака. Несмотря на то, что для кон-шемовских орбиталей теорема Купманса не применима, их энергию допустимо использовать для выводов качественного характера.
![]() В Таблице 2 представлены атомные заряды, рассчитанные в рамках анализа натуральных орбиталей (NPA) [23,24] методом B3LYP для комплексов [Fe(CN)6]3–/4– в газовой фазе и с учетом эффектов сольватации (PCM). Как видно из таблицы, при восстановлении [Fe(CN)6]3– электрон переносится главным образом на орбитали атомов N и Fe. Следует отметить, что учет сольватации комплексных ионов не приводит к заметному изменению в зарядовом распределении. Квантово-химические расчеты компонент тензора поляризуемости реагентов вдоль аксиального и экваториального направлений дают близкие величины ~0.12–0.13 нм3. Это означает, что в поле двойного слоя зарядовые распределения в частицах практически не меняются.
В Таблице 2 представлены атомные заряды, рассчитанные в рамках анализа натуральных орбиталей (NPA) [23,24] методом B3LYP для комплексов [Fe(CN)6]3–/4– в газовой фазе и с учетом эффектов сольватации (PCM). Как видно из таблицы, при восстановлении [Fe(CN)6]3– электрон переносится главным образом на орбитали атомов N и Fe. Следует отметить, что учет сольватации комплексных ионов не приводит к заметному изменению в зарядовом распределении. Квантово-химические расчеты компонент тензора поляризуемости реагентов вдоль аксиального и экваториального направлений дают близкие величины ~0.12–0.13 нм3. Это означает, что в поле двойного слоя зарядовые распределения в частицах практически не меняются.
![]() Важно отметить, что изменение длины связи Fe–C при переходе от окисленной к восстановленной форме изолированных комплексов не превышает 0.004 нм (Таблица 1), что сопоставимо с экспериментальными данными (0.002¸0.003 нм) [21]. На основе результатов, полученных методом B3LYP с использованием методики [25], были проведены расчеты внутрисферной энергии реорганизации lin комплексных реагентов. Для величины lin процесса восстановления получено значение 9.61 кДж•моль–1, а для обратной реакции 4.18 кДж•моль–1. Полученные результаты находятся в хорошем согласии с оценкой, приведенной в [21] (»4.18 кДж•моль–1). Таким образом, внутрисферная составляющая энергии реорганизации в реакциях восстановления/окисления [Fe(CN)6]3–/4–, по-видимому, не дает заметного вклада в энергию активации.
Важно отметить, что изменение длины связи Fe–C при переходе от окисленной к восстановленной форме изолированных комплексов не превышает 0.004 нм (Таблица 1), что сопоставимо с экспериментальными данными (0.002¸0.003 нм) [21]. На основе результатов, полученных методом B3LYP с использованием методики [25], были проведены расчеты внутрисферной энергии реорганизации lin комплексных реагентов. Для величины lin процесса восстановления получено значение 9.61 кДж•моль–1, а для обратной реакции 4.18 кДж•моль–1. Полученные результаты находятся в хорошем согласии с оценкой, приведенной в [21] (»4.18 кДж•моль–1). Таким образом, внутрисферная составляющая энергии реорганизации в реакциях восстановления/окисления [Fe(CN)6]3–/4–, по-видимому, не дает заметного вклада в энергию активации.
![]() Расчеты предсказывают незначительное увеличение длины связи C–N (»0.001нм) при переносе электрона на комплекс [Fe(CN)6]3– (см. Таблицу 1). Несмотря на то, что частота колебания группы CN лежит в квантовом диапазоне (2122 см–1 >> kT ), соответствующее значение фактора туннелирования exp(s) близко к 1.
Расчеты предсказывают незначительное увеличение длины связи C–N (»0.001нм) при переносе электрона на комплекс [Fe(CN)6]3– (см. Таблицу 1). Несмотря на то, что частота колебания группы CN лежит в квантовом диапазоне (2122 см–1 >> kT ), соответствующее значение фактора туннелирования exp(s) близко к 1.
![]() В Таблице 3 приведены результаты расчетов свободных энергий гидратации комплексов [Fe(CN)6]3–/4–, проведенных методом B3LYP с использованием модели PCM. В рамках данного подхода суммарная свободная энергия гидратации включает электростатическую часть (DGelst) и некулоновский вклад (дисперсионная поправка, энергия образования полости в растворителе и др.). Зная величину DGelst и используя известную формулу Борна, можно оценить эффективный радиус реагента (reff):
В Таблице 3 приведены результаты расчетов свободных энергий гидратации комплексов [Fe(CN)6]3–/4–, проведенных методом B3LYP с использованием модели PCM. В рамках данного подхода суммарная свободная энергия гидратации включает электростатическую часть (DGelst) и некулоновский вклад (дисперсионная поправка, энергия образования полости в растворителе и др.). Зная величину DGelst и используя известную формулу Борна, можно оценить эффективный радиус реагента (reff):
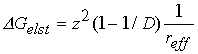 | (1) |
где z — заряд иона и D — статическая диэлектрическая проницаемости растворителя.
![]() Как видно из Таблицы 3, значения reff, рассчитанные таким образом для комплексов [Fe(CN)6]3–/4–, совпадают и составляют 0.41 нм. Полученный результат в целом сопоставим с оценкой, приведенной в [14] (»0.44 нм) и может быть использован для расчета внешнесферной энергии реорганизации (lout) редокс-процессов с участием [Fe(CN)6]3–/4–: l по простейшей формуле Маркуса с использованием оговоренного выше радиуса эффективной сферы даёт значение ~ 70 кДж/моль.
Как видно из Таблицы 3, значения reff, рассчитанные таким образом для комплексов [Fe(CN)6]3–/4–, совпадают и составляют 0.41 нм. Полученный результат в целом сопоставим с оценкой, приведенной в [14] (»0.44 нм) и может быть использован для расчета внешнесферной энергии реорганизации (lout) редокс-процессов с участием [Fe(CN)6]3–/4–: l по простейшей формуле Маркуса с использованием оговоренного выше радиуса эффективной сферы даёт значение ~ 70 кДж/моль.
![]() По независимой оценке из данных по кинетике гомогенных процессов [26], полная энергия реорганизации lout составляет ~90 кДж/моль, то есть несколько больше, чем сумма lin и lout , оцененных в этом разделе. Ниже при расчётах предполагается, что величина l лежит в пределах 70–90 кДж/моль, поэтому сопоставляются результаты для двух крайних значений этого интервала.
По независимой оценке из данных по кинетике гомогенных процессов [26], полная энергия реорганизации lout составляет ~90 кДж/моль, то есть несколько больше, чем сумма lin и lout , оцененных в этом разделе. Ниже при расчётах предполагается, что величина l лежит в пределах 70–90 кДж/моль, поэтому сопоставляются результаты для двух крайних значений этого интервала.
![]() Как было показано в [25], полиатомные реагенты со сложным распределеним заряда по объёму частицы могут быть аппроксимированы точечными реагентами на основе расчётов микроскопических работ сближения Wi (для реагентов) и Wf (для продуктов).
Как было показано в [25], полиатомные реагенты со сложным распределеним заряда по объёму частицы могут быть аппроксимированы точечными реагентами на основе расчётов микроскопических работ сближения Wi (для реагентов) и Wf (для продуктов).
![]() Расчёты величин Wi и Wf произведены по данным Таблиц 1 и 2 с использованием уравнения теории Гуи-Чапмана для потенциала yx на расстоянии x от внешней плоскости Гельмгольца [27]:
Расчёты величин Wi и Wf произведены по данным Таблиц 1 и 2 с использованием уравнения теории Гуи-Чапмана для потенциала yx на расстоянии x от внешней плоскости Гельмгольца [27]:
 | (2) |
где ![]() — обратная величина дебаевского радиуса. При этом, как и в [25], предполагалось, что точечные заряды жёстко закреплены на расстояниях xi от внешней плоскости Гельмгольца, определяемых геометрией частиц и условием нахождения частицы в диффузной части двойного слоя. Последнее означает, что расстояние максимального приближения соответствует касанию внешней плоскости Гельмгольца и сферы, в которую вписан октаэдрический комплексный ион(Основанием для предположения о том, что частица реагента полностью находится в диффузной части двойного слоя, являются данные [28] по специфической адсорбции [Fe(CN)6]4–. В области отрицательных зарядов электрода, для которой ниже обсуждаются экспериментальные данные, специфическая адсорбция отсутствует.). Зависимость Wi от yo (и, соответственно, от потенциала электрода) сопоставлена с рассчитанными для точечных зарядов, смещённых на разные расстояния от внешней плоскости Гельмгольца, на рис.1б. Аналогичные зависимости получаются и для других концентраций электролита фона; как Wi, так и Wf близки к соответствующим работам сближения точечного реагента, смещённого в объём раствора на x=0.42–0.45 нм, т.е. на величину, близкую к радиусу реагента и продукта [21].
— обратная величина дебаевского радиуса. При этом, как и в [25], предполагалось, что точечные заряды жёстко закреплены на расстояниях xi от внешней плоскости Гельмгольца, определяемых геометрией частиц и условием нахождения частицы в диффузной части двойного слоя. Последнее означает, что расстояние максимального приближения соответствует касанию внешней плоскости Гельмгольца и сферы, в которую вписан октаэдрический комплексный ион(Основанием для предположения о том, что частица реагента полностью находится в диффузной части двойного слоя, являются данные [28] по специфической адсорбции [Fe(CN)6]4–. В области отрицательных зарядов электрода, для которой ниже обсуждаются экспериментальные данные, специфическая адсорбция отсутствует.). Зависимость Wi от yo (и, соответственно, от потенциала электрода) сопоставлена с рассчитанными для точечных зарядов, смещённых на разные расстояния от внешней плоскости Гельмгольца, на рис.1б. Аналогичные зависимости получаются и для других концентраций электролита фона; как Wi, так и Wf близки к соответствующим работам сближения точечного реагента, смещённого в объём раствора на x=0.42–0.45 нм, т.е. на величину, близкую к радиусу реагента и продукта [21].
![]() Ранее в [5,6] обсуждалось влияние выбора значения yx на ход исправленных тафелевских зависимостей, построенных для эффективных точечных зарядов в соответствии с традиционной формулой:
Ранее в [5,6] обсуждалось влияние выбора значения yx на ход исправленных тафелевских зависимостей, построенных для эффективных точечных зарядов в соответствии с традиционной формулой:
| (3) |
(Здесь и далее величины z используются с учётом знака, а величины E приводятся в шкале нас. к. э., с — концентрация реагента K3[Fe(CN)6] в объеме раствора).
![]() На рис.2 (кривые 1–4) показаны ИТЗ для y=yo, кривые 1'–4' представляют собой ИТЗ, построенные с использованием рассчитанного по формуле (2) потенциала yx на расстоянии x=0.45 нм. Как видно из рисунка, поправка практически не влияет на степень совпадения кривых для разных концентраций электролита фона, но существенно изменяет наклон кривых и обеспечивает большую близость хода ИТЗ к линейному. Эффективное «отодвигание» реагента приводит к снижению кажущегося коэффициента переноса a (в области наиболее отрицательных потенциалов — до ~0,05), как это уже отмечалось ранее в [5].
На рис.2 (кривые 1–4) показаны ИТЗ для y=yo, кривые 1'–4' представляют собой ИТЗ, построенные с использованием рассчитанного по формуле (2) потенциала yx на расстоянии x=0.45 нм. Как видно из рисунка, поправка практически не влияет на степень совпадения кривых для разных концентраций электролита фона, но существенно изменяет наклон кривых и обеспечивает большую близость хода ИТЗ к линейному. Эффективное «отодвигание» реагента приводит к снижению кажущегося коэффициента переноса a (в области наиболее отрицательных потенциалов — до ~0,05), как это уже отмечалось ранее в [5].
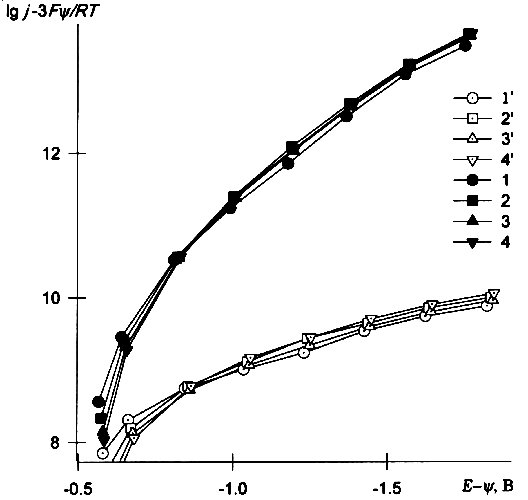 |
| Рис. 2. Исправленные тафелевские зависимости, построенные по данным [5,6] для растворов 0.33 мМ K3Fe(CN)6 + m мМ KСl по уравнению (2) с использованием значений y=yx для x: 1–4 — 0; 1'–4' — 0.45 нм; m: 1,1' — 0; 2,2' — 0.5; 3,3' — 1; 4,4' — 1.5. |
![]() Наиболее очевидным недостатком построения ИТЗ на основе уравнения (3), вне зависимости от типа yx-поправки, является игнорирование возможности участия в реакции ансамбля реагентов с различными зарядами.
Наиболее очевидным недостатком построения ИТЗ на основе уравнения (3), вне зависимости от типа yx-поправки, является игнорирование возможности участия в реакции ансамбля реагентов с различными зарядами.
![]() Из литературы известна величина константы ассоциации [Fe(CN)6]3– с катионом калия по первой ступени (
Из литературы известна величина константы ассоциации [Fe(CN)6]3– с катионом калия по первой ступени (![]() =1. 46 [29]). Она удовлетворительно согласуется с величиной, рассчитанной по уравнению Фоусса [30] при подстановке в него в качестве параметра межионного расстояния a величин 0.6–0.7 нм (
=1. 46 [29]). Она удовлетворительно согласуется с величиной, рассчитанной по уравнению Фоусса [30] при подстановке в него в качестве параметра межионного расстояния a величин 0.6–0.7 нм (![]() =1.3). Это позволяет использовать уравнение Фоусса и для оценки второй стадийной константы (процесса образования K2[Fe(CN)6]1–). При выборе для этого случая параметра а можно руководствоваться предположением о полной делокализации первого присоединённого катиона (тогда величина а такая же, как и для первой ступени). Если же межионное контактное расстояние для второй ступени ассоциации выше, то всего на 0.1–0.2 нм, что не вносит большой неточности: рассчитанные
=1.3). Это позволяет использовать уравнение Фоусса и для оценки второй стадийной константы (процесса образования K2[Fe(CN)6]1–). При выборе для этого случая параметра а можно руководствоваться предположением о полной делокализации первого присоединённого катиона (тогда величина а такая же, как и для первой ступени). Если же межионное контактное расстояние для второй ступени ассоциации выше, то всего на 0.1–0.2 нм, что не вносит большой неточности: рассчитанные ![]() лежат в любом случае в интервале 0.8–0.9.
лежат в любом случае в интервале 0.8–0.9.
![]() Указанные значения констант довольно велики и свидетельствуют о том, что в равновесных условиях в водном растворе K3[Fe(CN)6] даже в отсутствие фоновых ионов калия сосуществуют три формы анионов. Их относительные концентрации в объёме раствора, выраженные в процентах, для растворов 0.33 мМ K3[Fe(CN)6] с различными добавками электролита фона (растворы, использованные в [5,6]) представлены в Таблице 4.
Указанные значения констант довольно велики и свидетельствуют о том, что в равновесных условиях в водном растворе K3[Fe(CN)6] даже в отсутствие фоновых ионов калия сосуществуют три формы анионов. Их относительные концентрации в объёме раствора, выраженные в процентах, для растворов 0.33 мМ K3[Fe(CN)6] с различными добавками электролита фона (растворы, использованные в [5,6]) представлены в Таблице 4.
![]() Уравнение для тока при аддитивном участии реагентов разной зарядности имеет вид:
Уравнение для тока при аддитивном участии реагентов разной зарядности имеет вид:
 | (4) |
где z=–3 — заряд аниона [Fe(CN)6]3–; k и k' — отношения парциальных констант скоростей электровосстановления двухзарядного и однозарядного анионов к аналогичной константе для трёхзарядного; c1, c2 и c3 — объёмные концентрации трёхзарядного, двухзарядного и однозарядного анионов соответственно. Согласно этому уравнению, линеаризация экспериментальных поляризационных кривых может быть достигнута (в рамках теории замедленного разряда) в координатах, отличающихся от классических координат ИТЗ:
 | (5) |
Пример перестроенных таким образом ИТЗ для соотношений констант k=k'=1 и k=0, k'=0.1 показаны на рис.3а,б. Их наклоны при не слишком малых отрицательных потенциалах практически одинаковы, т.к. основной вклад в скорость изменения тока с потенциалом вносит анион с зарядом z=–1 (при различии констант не более чем на 1–3 порядка при k>0.001). ИТЗ для раствора без добавок фона (кривые 1, 1' рис. 3а, б) в области E>-1В имеют аномальный ход, что может быть, в принципе, связано с некорректностью использования в этом случае yx-потенциалов при не слишком больших отрицательных зарядах. Строго говоря, более вероятным является случай k¹k'¹1, поскольку расстояния переноса электрона для ассоциатов разного состава скорее всего различны. Попытки подбора величин k и k' при варьировании каждой из них в пределах 1–2 порядков не позволяют обеспечить ни линеаризации кривых, ни их совпадения для разных концентраций фона. В то же время наклон кривых, по крайней мере в области высоких потенциалов, мало зависит от выбора k и k' и оказывается значительно меньше, чем на рис.2. То, что в обсуждаемом интервале перенапряжений (Е – Е0) (Е0 — стандартный потенциал обсуждаемой редокс-системы) не удаётся получить постоянства a ни при каких разумных предположениях, не удивительно с учётом сделанного в [9] заключения о практически безактивационном характере процесса в области высоких перенапряжений.
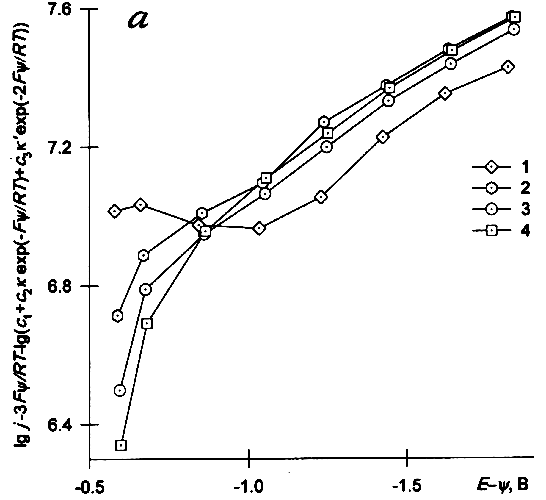 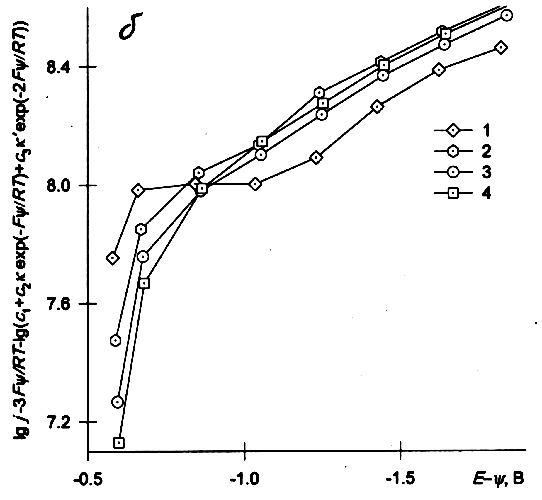 |
| Рис. 3. Исправленные тафелевские зависимости, построенные по данным [5,6] для растворов 0.33 мМ K3Fe(CN)6 + m мМ KСl по уравнению (4) с использованием значений y=yx для x=0.45 нм; m: 1,1' — 0; 2,2' — 0.5; 3,3' — 1; 4,4' — 1.5; а — k=1, k'=1, б — k=0, k'=0.1 |
![]() Действительно, оценивая a в рамках маркусовской теории по уравнению
Действительно, оценивая a в рамках маркусовской теории по уравнению
 |
для перенапряжений выше 1 В с учетом достаточно малых l получим формально отрицательные величины. Это, собственно, и является условием достижения ближайшей окрестности безактивационной области, и приводит к необходимости учитывать перенос электронов с нефермиевских уровней, компенсирующий снижение скорости процесса с перенапряжением в инвертированной маркусовской области.
![]() Теоретические величины коэффициента переноса a могут быть рассчитаны в этом случае по уравнению [31]:
Теоретические величины коэффициента переноса a могут быть рассчитаны в этом случае по уравнению [31]:
 | (6) |
где энергия e отсчитана от уровня Ферми, а интегрирование проводится по всем электронным уровням в металле.
![]() Сопоставляя значения a, найденные по построенным из экспериментальных данных ИТЗ в разных координатах, с их теоретическими значениями (рис.4 кривые 1' и 2'), можно отметить что как более высокие величины a, определённые по кривым 1–4 рис.2, так и меньшие, отвечающие кривым 1'–4' рис.2, выше теоретических во всём интервале (рис.4 кривые 1, 2). В то же время очевидно попадание a, полученных путём перестроения ИТЗ с использованием формул (2) и (5), в область теоретических значений в интервале потенциалов 0.6<½E – y½<1.1 B (рис.4 кривая 3). Более близки к теоретически рассчитанным величиныa, найденные для тех же соотношений констант (k'>0.05 и k<1) в области ½E – y½>1.1 B (рис.4 кривая 3). В последнем случае, однако, в эксперименте наблюдается крайне слабая зависимость a от потенциала и, как следствие, существенно превышающее теоретические значение a в области потенциалов ½E – y½~2 В. Необходимо проанализировать чувствительность этого результата к реальному строению реагентов-ассоциатов.
Сопоставляя значения a, найденные по построенным из экспериментальных данных ИТЗ в разных координатах, с их теоретическими значениями (рис.4 кривые 1' и 2'), можно отметить что как более высокие величины a, определённые по кривым 1–4 рис.2, так и меньшие, отвечающие кривым 1'–4' рис.2, выше теоретических во всём интервале (рис.4 кривые 1, 2). В то же время очевидно попадание a, полученных путём перестроения ИТЗ с использованием формул (2) и (5), в область теоретических значений в интервале потенциалов 0.6<½E – y½<1.1 B (рис.4 кривая 3). Более близки к теоретически рассчитанным величиныa, найденные для тех же соотношений констант (k'>0.05 и k<1) в области ½E – y½>1.1 B (рис.4 кривая 3). В последнем случае, однако, в эксперименте наблюдается крайне слабая зависимость a от потенциала и, как следствие, существенно превышающее теоретические значение a в области потенциалов ½E – y½~2 В. Необходимо проанализировать чувствительность этого результата к реальному строению реагентов-ассоциатов.
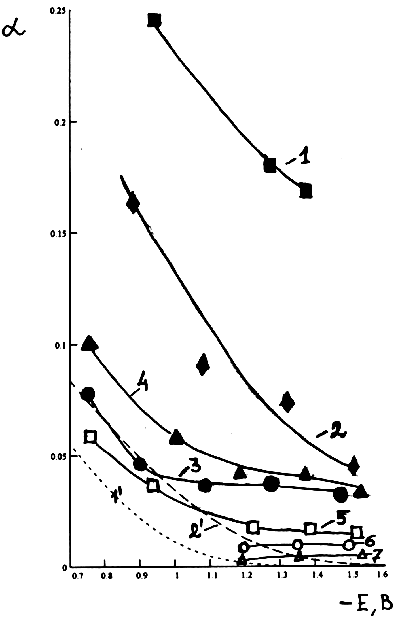 |
| Рис. 4. Зависимость величины коэффициента переноса электронов a от потенциала: 1 — определённая из кривой 3 рис.2, 2 —определённая из кривой 3' рис.2, 3 — определённая из кривой 3 рис.3; 4,5 — построенные с учётом микроскопических пси-прим-эффектов ассоциатов А и Б (4 — k''=0.01; 5 — k''=1); 6,7 — построенные с учётом микроскопических пси-прим-эффектов ассоциатов В, Г и Д (6 — k''(В)=1, k''(Г)= 0.1, k''(Д)=0.01; 7 — k''=1); 1', 2' — зависимости, рассчитанные по уравнению (5), l=: 1' — 70 кДж/моль, 2' — 90 кДж/моль |
![]() Не вполне обоснованным приближением, введённым выше при анализе участия в процессе ионных ассоциатов, является рассмотрение каждого из них как точечного реагента. Для оценки возможных особенностей электростатических взаимодействий реальных ассоциатов ниже проведено моделирование микроскопических yx- эффектов для нескольких упрощённых конфигураций ионных ассоциатов (рис.5).
Не вполне обоснованным приближением, введённым выше при анализе участия в процессе ионных ассоциатов, является рассмотрение каждого из них как точечного реагента. Для оценки возможных особенностей электростатических взаимодействий реальных ассоциатов ниже проведено моделирование микроскопических yx- эффектов для нескольких упрощённых конфигураций ионных ассоциатов (рис.5).
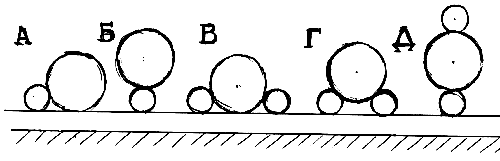 |
| Рис. 5. Схематические изображения разных конфигураций ионных ассоциатов в реакционном слое: большие круги — анионы, меньшие — катионы. |
![]() С целью качественного обсуждения проблемы влияния ионной ассоциации на строение реагента методом B3LYP былo исследовано строение ионной пары [Fe(CN)6]3–.... Na+. Оптимальная геометрия такого ассоциата использовалась в дальнейших расчетах в рамках модели PCM, что частично компенсирует неучет гидратной оболочки иона Na+. Как показал анализ атомных зарядов в ионном ассоциате, перенос заряда на Na+ пренебрежимо мал. В то же время Na+ вызывает некоторую асимметрию зарядового распределения в комплексе [Fe(CN)6]3– (см. Таблицы 5, 6), крайне незначительную в смысле изменений величины Wi. Поэтому при дальнейшем рассмотрении полагали, что как анионы, так и катионы можно аппроксимировать проводящими сферами с целочисленными зарядами, причём анионы, как и выше, считали сферами с радиусом около 0.45 нм. Выбор иона Na+ для упомянутых выше оценочных расчетов был обусловлен некоторыми методическими обстоятельствами, на качественном уровне все заключения можно распространить и на случай ассоциации с катионами калия.
С целью качественного обсуждения проблемы влияния ионной ассоциации на строение реагента методом B3LYP былo исследовано строение ионной пары [Fe(CN)6]3–.... Na+. Оптимальная геометрия такого ассоциата использовалась в дальнейших расчетах в рамках модели PCM, что частично компенсирует неучет гидратной оболочки иона Na+. Как показал анализ атомных зарядов в ионном ассоциате, перенос заряда на Na+ пренебрежимо мал. В то же время Na+ вызывает некоторую асимметрию зарядового распределения в комплексе [Fe(CN)6]3– (см. Таблицы 5, 6), крайне незначительную в смысле изменений величины Wi. Поэтому при дальнейшем рассмотрении полагали, что как анионы, так и катионы можно аппроксимировать проводящими сферами с целочисленными зарядами, причём анионы, как и выше, считали сферами с радиусом около 0.45 нм. Выбор иона Na+ для упомянутых выше оценочных расчетов был обусловлен некоторыми методическими обстоятельствами, на качественном уровне все заключения можно распространить и на случай ассоциации с катионами калия.
![]() Радиус гидратированного катиона при дальнейших оценках принимали равным 0.2 нм. Выражение для работ приближения реагента в этих конфигурациях приравнивали к работам приближения для эффективного точечного реагента и вычисляли (yx)eff по методике [32]:
Радиус гидратированного катиона при дальнейших оценках принимали равным 0.2 нм. Выражение для работ приближения реагента в этих конфигурациях приравнивали к работам приближения для эффективного точечного реагента и вычисляли (yx)eff по методике [32]:
| (7) |
где z=![]() =–2 для конфигураций А и Б рис.5 и –1 для остальных конфигураций.
=–2 для конфигураций А и Б рис.5 и –1 для остальных конфигураций.
![]() На рис.6 построены кривые зависимости (yx)eff от yo для различных ассоциатов, отвечающие общей концентрации K+ 2.5 мМ. В случае ассоциатов В и Д при |yo|>0.17 B величина (yx)eff незначительно меняется с ростом потенциала внешней плоскости Гельмгольца. Для ассоциата Г (yx)eff изменяется необычным образом, уменьшаясь по абсолютной величине с ростом потенциала yo. Сопоставляя рис.1 и 6, можно заключить, что при сильно выраженной неравномерности распределения заряда аппроксимация реагента точечным зарядом, смещённым на фиксированное расстояние, становится в широкой области потенциалов невозможной. Формально кривые рис. 6 соответствуют всё большему удалению реагента от электрода с ростом перенапряжения.
На рис.6 построены кривые зависимости (yx)eff от yo для различных ассоциатов, отвечающие общей концентрации K+ 2.5 мМ. В случае ассоциатов В и Д при |yo|>0.17 B величина (yx)eff незначительно меняется с ростом потенциала внешней плоскости Гельмгольца. Для ассоциата Г (yx)eff изменяется необычным образом, уменьшаясь по абсолютной величине с ростом потенциала yo. Сопоставляя рис.1 и 6, можно заключить, что при сильно выраженной неравномерности распределения заряда аппроксимация реагента точечным зарядом, смещённым на фиксированное расстояние, становится в широкой области потенциалов невозможной. Формально кривые рис. 6 соответствуют всё большему удалению реагента от электрода с ростом перенапряжения.
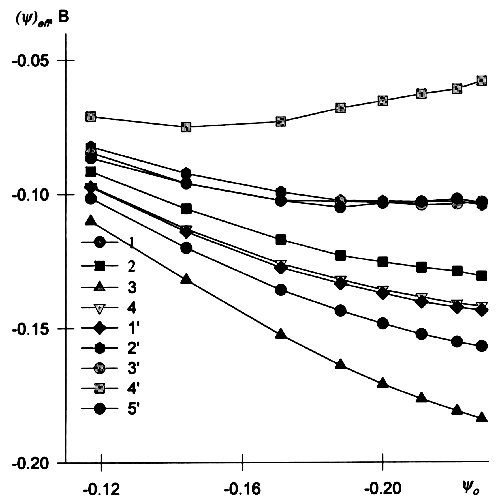 |
| Рис. 6. Зависимости (yx)eff, рассчитанным по уравнениям (6), от потенциала для различных конфигураций ассоциатов: 1' — 1, 2'— 2, 3' — 3, 4' — 4, 5' — 5 и, приведённые для сравнения, y=yx для х, нм: 1 — 0.45, 2 — 0.85, 3 — 0.2, 4 — 0.65. |
![]() Интересно отметить, что качественно близкий результат следует из формальной модели двойного слоя с двумя плоскостями приближения катионов [33]. Разумеется, в будущем необходима детализация таких расчётов на основе анализа реального строения гидратированного катиона.
Интересно отметить, что качественно близкий результат следует из формальной модели двойного слоя с двумя плоскостями приближения катионов [33]. Разумеется, в будущем необходима детализация таких расчётов на основе анализа реального строения гидратированного катиона.
![]() Подчеркнём, что обсуждаемые (yx)eff относятся к ионным парам, сформировавшимся в объёме раствора, и не могут быть использованы для анализа электростатических эффектов в случае дополнительной ассоциации вблизи межфазной границы. Ниже сделана попытка обработать имеющиеся экспериментальные данные с учётом оценок микроскопических пси-прим-эффектов для реагентов-ассоциатов.
Подчеркнём, что обсуждаемые (yx)eff относятся к ионным парам, сформировавшимся в объёме раствора, и не могут быть использованы для анализа электростатических эффектов в случае дополнительной ассоциации вблизи межфазной границы. Ниже сделана попытка обработать имеющиеся экспериментальные данные с учётом оценок микроскопических пси-прим-эффектов для реагентов-ассоциатов.
![]() Имеющихся экспериментальных данных, к сожалению, недостаточно для установления точного строения реакционного слоя. Однако можно провести некоторые оценки, исключающие из рассмотрения те или иные конфигурации. Как показано ниже, высокой чувствительностью к строению реакционного слоя обладает величина энергии активации в области высоких перенапряжений, т.к. из-за близости к безактивационной области значительный вклад в неё вносят величины Wi и Wf.
Имеющихся экспериментальных данных, к сожалению, недостаточно для установления точного строения реакционного слоя. Однако можно провести некоторые оценки, исключающие из рассмотрения те или иные конфигурации. Как показано ниже, высокой чувствительностью к строению реакционного слоя обладает величина энергии активации в области высоких перенапряжений, т.к. из-за близости к безактивационной области значительный вклад в неё вносят величины Wi и Wf.
![]() Учитывая локальное значение yx= (yx)eff, найдём изменение скорости электровосстановления ассоциатов 1–5 и аниона [Fe(CN)6]3– при возрастании температуры от 273 до 303К, для l, равных 70 и 90 кДж/моль и E=–1.2 В. Для этого ток при заданной температуре будем рассчитывать по формуле:
Учитывая локальное значение yx= (yx)eff, найдём изменение скорости электровосстановления ассоциатов 1–5 и аниона [Fe(CN)6]3– при возрастании температуры от 273 до 303К, для l, равных 70 и 90 кДж/моль и E=–1.2 В. Для этого ток при заданной температуре будем рассчитывать по формуле:
 | (8) |
где z — заряд ассоциата, а величина const включает слабо зависящую от e плотность электронов на уровне Ферми, трансмиссионный коэффициент, объемную концентрацию реагента и обычные константы [31].
![]() Относительное изменение тока в указанном температурном интервале будем сравнивать с соответствующим экспериментальным результатом [11] (24%).
Относительное изменение тока в указанном температурном интервале будем сравнивать с соответствующим экспериментальным результатом [11] (24%).
![]() В Таблице 7 представлены полученные величины изменения скорости электровосстановления с температурой, выраженные в процентах, для раствора 0,33 мМ K3[Fe(CN)6] + 1.5 мМ KСl при потенциале –1.2В.
В Таблице 7 представлены полученные величины изменения скорости электровосстановления с температурой, выраженные в процентах, для раствора 0,33 мМ K3[Fe(CN)6] + 1.5 мМ KСl при потенциале –1.2В.
![]() Как видно из таблицы, только конфигурации на основе тройных ассоциатов позволяют получить близкие к эксперименту температурные зависимости. Это согласуется с предположением о преимущественном участии в реакции именно однозарядных частиц и позволяет в принципе отказаться от предположения о смене механизма процесса с температурой (в смысле перехода от более быстрого мостикового к обычному переносу электрона).
Как видно из таблицы, только конфигурации на основе тройных ассоциатов позволяют получить близкие к эксперименту температурные зависимости. Это согласуется с предположением о преимущественном участии в реакции именно однозарядных частиц и позволяет в принципе отказаться от предположения о смене механизма процесса с температурой (в смысле перехода от более быстрого мостикового к обычному переносу электрона).
![]() Одним из важнейших качественных аргументов в пользу мостикового механизма процесса в литературе считается наличие существенной зависимости скорости восстановления феррицианида от природы присутствующего в системе катиона. Оценки, выполненные по уравнению (7) для модельных катионов с размерами 0.15 и 0.25 нм (что приблизительно соответствует радиусам гидратированных ионнов Сs+ и Li+), показывают, что скорость процесса при переходе от Li+ к Сs+ за счёт выигрыша в электростатической составляющей энергии активации может увеличиться всего в 1.9–2.7 раза для однозарядных ассоциатов. Этот результат мало зависит от конфигурации в реакционном слое. Ожидаемый эффект несколько ослабляется при уменьшении концентрации. Аналогичные оценки для ионных пар с зарядом –2 приводят к более слабым эффектам (увеличение всего в 1.3–1.6 раза).
Одним из важнейших качественных аргументов в пользу мостикового механизма процесса в литературе считается наличие существенной зависимости скорости восстановления феррицианида от природы присутствующего в системе катиона. Оценки, выполненные по уравнению (7) для модельных катионов с размерами 0.15 и 0.25 нм (что приблизительно соответствует радиусам гидратированных ионнов Сs+ и Li+), показывают, что скорость процесса при переходе от Li+ к Сs+ за счёт выигрыша в электростатической составляющей энергии активации может увеличиться всего в 1.9–2.7 раза для однозарядных ассоциатов. Этот результат мало зависит от конфигурации в реакционном слое. Ожидаемый эффект несколько ослабляется при уменьшении концентрации. Аналогичные оценки для ионных пар с зарядом –2 приводят к более слабым эффектам (увеличение всего в 1.3–1.6 раза).
![]() Значительная зависимость от природы катиона, наблюдаемая в эксперименте [5,6] (рост тока почти на порядок при переходе от Li+ к Сs+), не может быть обусловлена только электростатическими причинами. Дополнительное усиление этого эффекта обеспечивается, по-видимому, уменьшением расстояния переноса электрона при переходе от Li+ к Сs+. Нельзя исключить также увеличения долевого содержания тройных ассоциатов в объёме раствора. Таким образом, эффект влияния катиона имеет сложную природу, которая в дальнейшем требует специального исследования.
Значительная зависимость от природы катиона, наблюдаемая в эксперименте [5,6] (рост тока почти на порядок при переходе от Li+ к Сs+), не может быть обусловлена только электростатическими причинами. Дополнительное усиление этого эффекта обеспечивается, по-видимому, уменьшением расстояния переноса электрона при переходе от Li+ к Сs+. Нельзя исключить также увеличения долевого содержания тройных ассоциатов в объёме раствора. Таким образом, эффект влияния катиона имеет сложную природу, которая в дальнейшем требует специального исследования.
![]() Оценим долевые содержания ионных ассоциатов ni в реакционном слое на основе соотношения [25]:
Оценим долевые содержания ионных ассоциатов ni в реакционном слое на основе соотношения [25]:
| (9) |
где ck — объёмная доля k-го ассоциата (Таблица 4). При расчете cчитали, что в объеме раствора частицы одинаковой зарядности, образующие разные конфигурации в реакционном слое, неразличимы (основанием для этого являются малые характерные времена обмена катионами между ионными парами и объемом раствора). Поэтому в качестве ck для ассоциатов А и Б (и, соответственно, для В, Г и Д) подставляли одни и те же величины. В Таблице 8 представлены полученные при подстановке Wi=zF(yx)eff величины мольных долей в реакционном слое для различных конфигураций ассоциатов при разных потенциалах для раствора 0.33 мМ K3[Fe(CN)6] + 1.5 мМ KСl.
![]() Как видно из Таблицы 8, в реакционном слое преобладают ассоциаты а не трёхзарядные анионы. На рис. 7 показаны кривые изменения ni с ростом потенциала для таких ассоциатов.
Как видно из Таблицы 8, в реакционном слое преобладают ассоциаты а не трёхзарядные анионы. На рис. 7 показаны кривые изменения ni с ростом потенциала для таких ассоциатов.
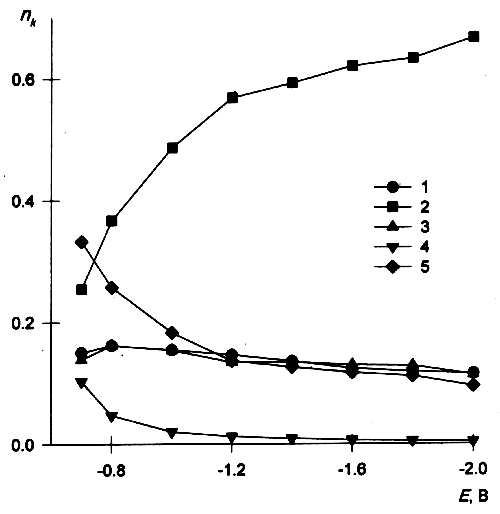 |
| Рис. 7. Зависимости nk от потенциала для конфигураций ассоциатов реагента: 1 — В, 2 — Г, 3 — Д; 4 — А, 5 — Б (в соответствии с обозначениями на рис. 5). |
![]() Исходя из выше сказанного и предполагая, что при различии расстояний туннелирования не более чем на 0.1–0.2 нм, парциальные константы скоростей восстановления разных ассоциатов будут отличаться не более, чем на 1–2 порядка в области потенциалов ½Е – y½>1 В, перестроим ИТЗ по формуле, аналогичной (5):
Исходя из выше сказанного и предполагая, что при различии расстояний туннелирования не более чем на 0.1–0.2 нм, парциальные константы скоростей восстановления разных ассоциатов будут отличаться не более, чем на 1–2 порядка в области потенциалов ½Е – y½>1 В, перестроим ИТЗ по формуле, аналогичной (5):
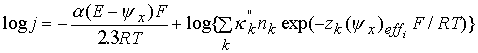 | (10) |
где k''-отношения парциальных констант скорости восстановления ассоциатов А, Б, В, Г, Д к парциальной константе скорости ассоциата А.
![]() На рис.8 представлены ИТЗ, построенные для различных k'' для случаев участия только двухзарядных анионов (z=–2) (кривые 1, 2) и однозарядных анионов (z=–1) (кривые 3, 4).
На рис.8 представлены ИТЗ, построенные для различных k'' для случаев участия только двухзарядных анионов (z=–2) (кривые 1, 2) и однозарядных анионов (z=–1) (кривые 3, 4).
 |
| Рис. 8. Исправленные тафелевские зависимости, построенные по данным [5,6] для растворов 0.33 мМ K3Fe(CN)6 + m мМ KСl по уравнению (8) с использованием значений (yx)eff рассчитанным по уравнениям (6) для конфигураций ассоциатов А и Б: 1- k''=0.01; 2- k''=1; для конфигураций ассоциатов В, Г, Д: 3-k''(В)=1, k''(Г)= 0.1, k''(Д)=0.01; 4- k''=1. |
![]() При использовании уравнений типа (3), (4) в микроскопической версии необходимо учесть, что величины (yx)eff, определяемые работой приближения реагента, могут быть использованы только в одном слагаемом ( его величина откладывается в комбинации с lg j на оси ординат). Величина E на оси абсцисс определяется выражением для свободной энергии реакции [(E – E0)F + Wi – Wf), и соответствующие эффективные (yx)'eff = (Wi – Wf )/Fz (для этой реакции они равны yx, где х — расстояние от плоскости максимального приближения до центра восстановления аниона) в общем случае отличаются от (yx)eff [32]. Для рассмотренных конфигураций разница (yx)'eff не слишком велика (см. рис. 6 кривые 1, 2, 4): максимальный сдвиг ИТЗ по оси абсцисс составит ~50мВ и к существенным изменениям не приведёт. Поэтому можно ограничиться использованием среднего значения.
При использовании уравнений типа (3), (4) в микроскопической версии необходимо учесть, что величины (yx)eff, определяемые работой приближения реагента, могут быть использованы только в одном слагаемом ( его величина откладывается в комбинации с lg j на оси ординат). Величина E на оси абсцисс определяется выражением для свободной энергии реакции [(E – E0)F + Wi – Wf), и соответствующие эффективные (yx)'eff = (Wi – Wf )/Fz (для этой реакции они равны yx, где х — расстояние от плоскости максимального приближения до центра восстановления аниона) в общем случае отличаются от (yx)eff [32]. Для рассмотренных конфигураций разница (yx)'eff не слишком велика (см. рис. 6 кривые 1, 2, 4): максимальный сдвиг ИТЗ по оси абсцисс составит ~50мВ и к существенным изменениям не приведёт. Поэтому можно ограничиться использованием среднего значения.
![]() Ход ИТЗ рис. 8 в области менее высоких перенапряжений (E=-0.7- -1 В) при введении микроскопических yx поправок является нормальным только при рассмотреннии электровосстановления двухзарядного аниона, и аномальным (формально отвечает отрицательным a) при рассмотрении электровосстановления только однозарядного аниона.
Ход ИТЗ рис. 8 в области менее высоких перенапряжений (E=-0.7- -1 В) при введении микроскопических yx поправок является нормальным только при рассмотреннии электровосстановления двухзарядного аниона, и аномальным (формально отвечает отрицательным a) при рассмотрении электровосстановления только однозарядного аниона.
![]() Основной особенностью всех кривых рис. 8 (по сравнению с ИТЗ, построенными разными способами для точечных реагентов) являются более низкие наклоны в области E= –1 — –2 В. Они отвечают a ~ 0.008 для однозарядного аниона и a ~ 0.016 для двухзарядного, т.е. значительно лучше согласуются с теоретическими значениями в этом интервале (<0.02), чем a, полученные в предыдущих построениях (рис. 4 кривые 4, 5).
Основной особенностью всех кривых рис. 8 (по сравнению с ИТЗ, построенными разными способами для точечных реагентов) являются более низкие наклоны в области E= –1 — –2 В. Они отвечают a ~ 0.008 для однозарядного аниона и a ~ 0.016 для двухзарядного, т.е. значительно лучше согласуются с теоретическими значениями в этом интервале (<0.02), чем a, полученные в предыдущих построениях (рис. 4 кривые 4, 5).
![]() На основании проведённого анализа нельзя полностью исключить эффект образования ионных ассоциатов непосредственно в реакционном слое. Это явление требует специального теоретического анализа. В то же время не проводившийся ранее учёт объёмных свойств растворов феррицианида и достаточно грубые оценки микроскопического строения ионных ассоциатов показывают, что необходимо рассматривать в первую очередь реакции с участием ассоциатов-тройников, а не самих анионов и ионных пар. Это позволяет даже без учёта локальных взаимодействий получить из эксперимента близкие к теоретическим величины коэффициента переноса и объяснить низкие значения энергии активации, то есть обосновать юлизкий к безактивационному характер процесса.
На основании проведённого анализа нельзя полностью исключить эффект образования ионных ассоциатов непосредственно в реакционном слое. Это явление требует специального теоретического анализа. В то же время не проводившийся ранее учёт объёмных свойств растворов феррицианида и достаточно грубые оценки микроскопического строения ионных ассоциатов показывают, что необходимо рассматривать в первую очередь реакции с участием ассоциатов-тройников, а не самих анионов и ионных пар. Это позволяет даже без учёта локальных взаимодействий получить из эксперимента близкие к теоретическим величины коэффициента переноса и объяснить низкие значения энергии активации, то есть обосновать юлизкий к безактивационному характер процесса.
![]() Зависимости a от потенциала, полученные из различных построений ИТЗ, могут отличаться по форме от теоретических по крайней мере из-за двух эффектов. Во-первых, это, возможно, связано с резким повышением концентрации ионов калия вблизи плоскости максимального приближения и образованием дополнительных ионных пар в реакционном слое. Именно такое предположение было обосновано на качественном уровне ранее [6]. Во-вторых, возможен обратный процесс разрушения подошедших из объёма ионных пар в сильном электрическом поле двойного слоя.
Зависимости a от потенциала, полученные из различных построений ИТЗ, могут отличаться по форме от теоретических по крайней мере из-за двух эффектов. Во-первых, это, возможно, связано с резким повышением концентрации ионов калия вблизи плоскости максимального приближения и образованием дополнительных ионных пар в реакционном слое. Именно такое предположение было обосновано на качественном уровне ранее [6]. Во-вторых, возможен обратный процесс разрушения подошедших из объёма ионных пар в сильном электрическом поле двойного слоя.
![]() Обоснованное выше заключение о безактивационном характере восстановления феррицианида не является частным результатом. Впервые на примере этой реакции можно рассмотреть закономерности перехода от обычного к безактивационному разряду, которые ранее не анализировались. В отличие от аналогичного перехода к безбарьерному разряду, в данном случае предсказываемые теоретически плавные изменения коэффициента переноса подтверждаются на опыте.
Обоснованное выше заключение о безактивационном характере восстановления феррицианида не является частным результатом. Впервые на примере этой реакции можно рассмотреть закономерности перехода от обычного к безактивационному разряду, которые ранее не анализировались. В отличие от аналогичного перехода к безбарьерному разряду, в данном случае предсказываемые теоретически плавные изменения коэффициента переноса подтверждаются на опыте.
![]() Работа выполнена при поддержке РФФИ, проект 99–03–32367а.
Работа выполнена при поддержке РФФИ, проект 99–03–32367а.
| комплекс | метод | r(Fe–Cax) | r(Fe–Ceq) | r(Cax–Nax) | r(Ceq–Neq) |
| [Fe(CN)6]3– | SCF B3LYP B3LYP (6-31g (d)) |
0.206401 0.19811 0.19851 |
0.211883 0.1984 0.19968 |
0.115001 0.119724 0.11763 |
0.116243 0.11961 0.11757 |
| [Fe(CN)6]4– | SCF MP2 B3LYP |
0.23224 0.19944 0.2026 |
0.23244 0.29944 0.2026 |
0.11812 0.12379 0.12088 |
0.11812 0.12379 0.12088 |
| z(Fe) | z(Cax) | z(Ceq) | z(Nax) | z(Neq) | |
| [Fe(CN)6]3– | +0.171 | +0.108 | +0.108 | –0.657 | –0.627 |
| +0.l83 | +0.081 | +0.084 | –0.633 | –0.627 | |
| [Fe(CN)6]4– | –0.18 | +0.138 | +0.138 | –0.774 | –0.775 |
| –0.15 | +0.083 | +0.083 | –0.72 | –0.731 |
| Комплекс | –DGsolv, кДж/моль |
(–DGelst), кДж/моль |
reff, нм |
| [Fe(CN)6]3– | 1441 | 1492 | 0.41 |
| [Fe(CN)6]4– | 2610 | 2660 | 0.41 |
| m | доля,% | ||
| [Fe(CN)6]3– | K[Fe(CN)6]2– | K2[Fe(CN)6]1– | |
| 0.0 | 97.2 | 2.78 | 0.02 |
| 0.5 | 95.8 | 4.15 | 0.05 |
| 1 | 94.6 | 5.31 | 0.09 |
| 1.5 | 93.4 | 6.5 | 0.1 |
| r(Fe–Cax) | r(Fe–Ceq) | r(Cax–Nax) | r(Ceq–Neq) | r(Fe–Na) |
| 0.19308 | 0.19721 | 0.11961 | 0.11941 | 0.52003 |
| z(Fe) | z(Cax)* | z(Cax) | z(Ceq) | z(Nax)* | z(Nax) | z(Neq) | z(Na) |
| +0.127 | +0.16 | +0.096 | +0.09 | –0.69 | –0.6 | –0.6 | +0.98 |
| конфигурация |
возрастание скорости, % |
|
| l=70 кДж/моль | l=90 кДж/моль | |
| Fe(CN)63– | 666 | 658 |
| А | 227 | 224 |
| Б | 148 | 143 |
| В | 61.5 | 59 |
| Г | 39 | 37 |
| Д | 61.5 | 59 |
| Е, В | конфигурации ассоциатов | |||||
| Fe(CN)63– | А | Б | В | Г | Д | |
| –0.7 | 0.0201 | 0.1034 | 0.3327 | 0.1503 | 0.2544 | 0.1391 |
| –0.8 | 3.9•10–3 | 0.0473 | 0.2575 | 0.162 | 0.3673 | 0.162 |
| –1 | 7.5•10–4 | 0.0203 | 0.183 | 0.1548 | 0.4869 | 0.1542 |
| –1.2 | 2.9•10–4 | 0.0123 | 0.1376 | 0.1467 | 0.5693 | 0.1346 |
| –1.4 | 1.56•10–4 | 8.69•10–3 | 0.1264 | 0.1362 | 0.594 | 0.1346 |
| –1.6 | 9.3•10–5 | 6.38•10–3 | 0.1174 | 0.124 | 0.6223 | 0.1299 |
| –1.8 | 6.3•10–5 | 5.16•10–3 | 0.1117 | 0.1194 | 0.6352 | 0.1285 |
| –2 | 4.85•10–5 | 4.46•10–3 | 0.0954 | 0.116 | 0.6699 | 0.1142 |