![]() Изучено электровосстановление биядерного комплекса меди (II) с лигандом Робсоновского типа на основе 4-трет-бутил-2,6-диформилфенола и 1,3-диаминопропана в водной среде на капающем ртутном, золотом, амальгамированном золотом и стеклоуглеродном электродах. Данные экспериментов по препаративному электролизу, измерений методом циклической вольтамперометрии и классической полярографии говорят о том, что изучаемый комплекс восстанавливается в водной среде с переносом четырех электронов.
Изучено электровосстановление биядерного комплекса меди (II) с лигандом Робсоновского типа на основе 4-трет-бутил-2,6-диформилфенола и 1,3-диаминопропана в водной среде на капающем ртутном, золотом, амальгамированном золотом и стеклоуглеродном электродах. Данные экспериментов по препаративному электролизу, измерений методом циклической вольтамперометрии и классической полярографии говорят о том, что изучаемый комплекс восстанавливается в водной среде с переносом четырех электронов.
![]() Ключевые слова: биядерный комплекс, электровосстановление, полярография, препаративный электролиз.
Ключевые слова: биядерный комплекс, электровосстановление, полярография, препаративный электролиз.
![]() Макроциклические полидентатные лиганды, включающие основания Шиффа, были впервые синтезированы в 70-х годах [1]. Тогда же удалось [2,3] впервые получить биядерные комплексы ряда переходных металлов например, Cu, Ni, Zn, Co, Fe, Mn на основе таких лигандов (ниже - комплексы Робсоновского типа, КРТ). КРТ являются предметом разнообразных электрохимических исследований. В значительной степени это связано с попытками моделирования центров металлосодержащих ферментов [4,5] и с перспективными каталитическими приложениями. Среди последних специально следует отметить способность к активации малых молекул (например, CO, N2) [1,3,4], обусловленную образованием аддуктов [6]. Кроме того, исследования стимулируются поиском амперометрических сенсоров на основе разнообразных планарных комплексов с хелатирующими лигандами [7].
Макроциклические полидентатные лиганды, включающие основания Шиффа, были впервые синтезированы в 70-х годах [1]. Тогда же удалось [2,3] впервые получить биядерные комплексы ряда переходных металлов например, Cu, Ni, Zn, Co, Fe, Mn на основе таких лигандов (ниже - комплексы Робсоновского типа, КРТ). КРТ являются предметом разнообразных электрохимических исследований. В значительной степени это связано с попытками моделирования центров металлосодержащих ферментов [4,5] и с перспективными каталитическими приложениями. Среди последних специально следует отметить способность к активации малых молекул (например, CO, N2) [1,3,4], обусловленную образованием аддуктов [6]. Кроме того, исследования стимулируются поиском амперометрических сенсоров на основе разнообразных планарных комплексов с хелатирующими лигандами [7].
![]() Молекула КРТ построена так, что два центральных иона, находящиеся в окружении электронодонорных лигандов, расположены на близком расстоянии (0.311 – 0.315 нм для различных КРТ [5,8,9]), что обуславливает их существенное электронное перекрывание через мостиковые кислородные атомы [3,5]. Благодаря более низкой, чем для нециклических аналогов, лабильности биядерных комплексов с макроциклическими лигандами КРТ представляют собой удобные объекты для изучения кинетики внутримолекулярного переноса электрона между металлическими центрами и устойчивости соединений в смешанно-валентных состояниях [6].
Молекула КРТ построена так, что два центральных иона, находящиеся в окружении электронодонорных лигандов, расположены на близком расстоянии (0.311 – 0.315 нм для различных КРТ [5,8,9]), что обуславливает их существенное электронное перекрывание через мостиковые кислородные атомы [3,5]. Благодаря более низкой, чем для нециклических аналогов, лабильности биядерных комплексов с макроциклическими лигандами КРТ представляют собой удобные объекты для изучения кинетики внутримолекулярного переноса электрона между металлическими центрами и устойчивости соединений в смешанно-валентных состояниях [6].
![]() В некоторых отношениях можно усмотреть аналогию КРТ с порфириновыми и фталоцианиновыми комплексами (плоская геометрия, сопряженная система двойных связей, стабильность комплексов со многими переходными металлами, каталитические свойства) [10]. На основании этой аналогии можно предположить и склонность КРТ к адсорбции на электродах.
В некоторых отношениях можно усмотреть аналогию КРТ с порфириновыми и фталоцианиновыми комплексами (плоская геометрия, сопряженная система двойных связей, стабильность комплексов со многими переходными металлами, каталитические свойства) [10]. На основании этой аналогии можно предположить и склонность КРТ к адсорбции на электродах.
![]() Существенное отличие от N4-комплексов, позволяющее расширить круг исследуемых адсорбционных и каталитических явлений, обусловлено наличием двух редокс-центров. Эта же особенность химического строения может быть в перспективе использована в фундаментальных исследованиях элементарного акта многоэлектронных процессов, в частности, в связи с эффектами электронной корреляции [11]. Крайне привлекательной является, в частности, возможность исследовать влияние растворителя на закономерности последовательного электронного переноса.
Существенное отличие от N4-комплексов, позволяющее расширить круг исследуемых адсорбционных и каталитических явлений, обусловлено наличием двух редокс-центров. Эта же особенность химического строения может быть в перспективе использована в фундаментальных исследованиях элементарного акта многоэлектронных процессов, в частности, в связи с эффектами электронной корреляции [11]. Крайне привлекательной является, в частности, возможность исследовать влияние растворителя на закономерности последовательного электронного переноса.
![]() Основная масса литературных данных по электрохимии КРТ относится к их восстановлению или окислению в органических средах [5,6,9,12-15]. В то же время данные по электровосстановлению КРТ в водной среде практически отсутствуют, возможно, в силу ограниченной растворимости большинства комплексов подобного типа. Реализация электродных превращений в водных растворах могла бы существенно расширить круг электрокаталитических систем. К настоящему времени наиболее подробно изученными КРТ являются, пожалуй, гомобиядерные комплексы меди с макроциклическими основаниями Шиффа на основе 4-замещенных-2,6-диформилфенолов и диаминов различного строения. В частности, на примере макроциклического биядерного комплекса меди с лигандом на основе 4-метил-2,6-диформилфенола и 1,3-диаминопропана удается наблюдать два последовательных одноэлектронных процесса с потенциалами полуволн, различающимися на 350 - 450 мВ в зависимости от природы органического растворителя [5,6,9,12,16,17]. Известно также [3], что в водной среде этот комплекс является 2:1 электролитом, образуя двухзарядный биядерный катион.
Основная масса литературных данных по электрохимии КРТ относится к их восстановлению или окислению в органических средах [5,6,9,12-15]. В то же время данные по электровосстановлению КРТ в водной среде практически отсутствуют, возможно, в силу ограниченной растворимости большинства комплексов подобного типа. Реализация электродных превращений в водных растворах могла бы существенно расширить круг электрокаталитических систем. К настоящему времени наиболее подробно изученными КРТ являются, пожалуй, гомобиядерные комплексы меди с макроциклическими основаниями Шиффа на основе 4-замещенных-2,6-диформилфенолов и диаминов различного строения. В частности, на примере макроциклического биядерного комплекса меди с лигандом на основе 4-метил-2,6-диформилфенола и 1,3-диаминопропана удается наблюдать два последовательных одноэлектронных процесса с потенциалами полуволн, различающимися на 350 - 450 мВ в зависимости от природы органического растворителя [5,6,9,12,16,17]. Известно также [3], что в водной среде этот комплекс является 2:1 электролитом, образуя двухзарядный биядерный катион.
![]() В настоящей работе предпринята попытка дать общую характеристику электрохимических свойств биядерного комплекса меди с лигандом Робсоновского типа на основе 4-трет-бутил-2,6-диформилфенола и 1,3-диаминопропана (рис.1), сокращенно обозначенного ниже [Cu2L]Cl2, в водных растворах. Выбор данного комплекса с внешнесферным хлорид-анионом обусловлен его достаточно высокой растворимостью в воде.
В настоящей работе предпринята попытка дать общую характеристику электрохимических свойств биядерного комплекса меди с лигандом Робсоновского типа на основе 4-трет-бутил-2,6-диформилфенола и 1,3-диаминопропана (рис.1), сокращенно обозначенного ниже [Cu2L]Cl2, в водных растворах. Выбор данного комплекса с внешнесферным хлорид-анионом обусловлен его достаточно высокой растворимостью в воде.
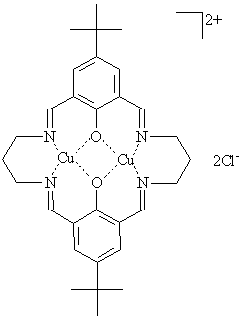 |
| Рис. 1. Схематическое изображение комплекса [Cu2L]Cl2. |
![]() Все измерения проводились с использованием 0,1 М ацетатных буферных растворов в качестве фоновых.
Все измерения проводились с использованием 0,1 М ацетатных буферных растворов в качестве фоновых.
![]() Реагент [Cu2L]Cl2·H2O, синтезированный по методике [18], очищали перекристаллизацией из воды (Milli-Q). 0.1 М буферные растворы готовили на дистиллированной воде, дополнительно очищенной на установке Milli-Q, из ацетата натрия марки хч (трижды очищенного перекристаллизацией) и ледяной уксусной кислоты (Merck). Раствор 20 мМ сульфата меди с добавкой 0.1 М ацетатной буферной смеси (ниже AcH + AcNa ) (pH 6.0) приготовлен из прокаленного CuSO4·6H2O. Для приготовления насыщенного раствора 4-трет-бутил-2,6-диформилфенола в 0.1 М ацетатном буфере (pH 6.0) использовали 4-трет-бутил-2,6-диформилфенол, синтезированный по методике [20].
Реагент [Cu2L]Cl2·H2O, синтезированный по методике [18], очищали перекристаллизацией из воды (Milli-Q). 0.1 М буферные растворы готовили на дистиллированной воде, дополнительно очищенной на установке Milli-Q, из ацетата натрия марки хч (трижды очищенного перекристаллизацией) и ледяной уксусной кислоты (Merck). Раствор 20 мМ сульфата меди с добавкой 0.1 М ацетатной буферной смеси (ниже AcH + AcNa ) (pH 6.0) приготовлен из прокаленного CuSO4·6H2O. Для приготовления насыщенного раствора 4-трет-бутил-2,6-диформилфенола в 0.1 М ацетатном буфере (pH 6.0) использовали 4-трет-бутил-2,6-диформилфенол, синтезированный по методике [20].
![]() Измерения pH проводили с использованием иономера универсального ЭВ-74.
Измерения pH проводили с использованием иономера универсального ЭВ-74.
![]() Во всех электрохимических измерениях использовали насыщенный каломельный электрод сравнения, соединенный с сосудом рабочего электрода солевым мостиком (насыщенный KCl). Все величины потенциалов приводятся ниже в шкале нас. к. э.
Во всех электрохимических измерениях использовали насыщенный каломельный электрод сравнения, соединенный с сосудом рабочего электрода солевым мостиком (насыщенный KCl). Все величины потенциалов приводятся ниже в шкале нас. к. э.
![]() Измерения в 0.3 мМ растворах реагента [Cu2L]Cl2 при pH 4 – 6 проводили методом классической полярографии. Вспомогательным электродом служила платиновая пластинка, расположенная в сосуде рабочего электрода. Скорость вытекания ртути составляла 0.784 мг/с, период капания 8 с при высоте ртутного столба 45 см для разомкнутой цепи в воде. В некоторых опытах высоту столба ртути варьировали от 45 до 72 см. Перед измерениями растворы деаэрировали пропусканием водорода в течение минимум 40 мин во вспомогательном сосуде, а затем еще около 15 мин в ячейке. Полученные кривые исправляли на измеренные в фоновых растворах токи заряжения, которые составляли не более 15% от суммарного тока.
Измерения в 0.3 мМ растворах реагента [Cu2L]Cl2 при pH 4 – 6 проводили методом классической полярографии. Вспомогательным электродом служила платиновая пластинка, расположенная в сосуде рабочего электрода. Скорость вытекания ртути составляла 0.784 мг/с, период капания 8 с при высоте ртутного столба 45 см для разомкнутой цепи в воде. В некоторых опытах высоту столба ртути варьировали от 45 до 72 см. Перед измерениями растворы деаэрировали пропусканием водорода в течение минимум 40 мин во вспомогательном сосуде, а затем еще около 15 мин в ячейке. Полученные кривые исправляли на измеренные в фоновых растворах токи заряжения, которые составляли не более 15% от суммарного тока.
![]() Потенциодинамические измерения проводили в трехэлектродной ячейке с разделенными пространствами и вспомогательным электродом из платиновой пластинки. В качестве рабочих электродов использовали стеклоуглеродную пластинку, амальгамированную золотую проволоку, золотую проволоку. Перед каждой серией измерений поверхность стеклоуглерода очищали механически стеклянным порошком и промывали водой. Золотой электрод предварительно подвергали травлению в кипящей царской водке в течение 10 - 30 с и затем тщательно промывали водой. Для приготовления амальгамы поверхность электрода очищали в концентрированной азотной кислоте, затем в кипящей царской водке, погружали в ртуть не менее, чем на 5 мин, а затем образовавшуюся поверхность амальгамы промывали водой. Перед каждой серией измерений растворы деаэрировали аргоном в течение 25 - 30 минут.
Потенциодинамические измерения проводили в трехэлектродной ячейке с разделенными пространствами и вспомогательным электродом из платиновой пластинки. В качестве рабочих электродов использовали стеклоуглеродную пластинку, амальгамированную золотую проволоку, золотую проволоку. Перед каждой серией измерений поверхность стеклоуглерода очищали механически стеклянным порошком и промывали водой. Золотой электрод предварительно подвергали травлению в кипящей царской водке в течение 10 - 30 с и затем тщательно промывали водой. Для приготовления амальгамы поверхность электрода очищали в концентрированной азотной кислоте, затем в кипящей царской водке, погружали в ртуть не менее, чем на 5 мин, а затем образовавшуюся поверхность амальгамы промывали водой. Перед каждой серией измерений растворы деаэрировали аргоном в течение 25 - 30 минут.
![]() Потенциодинамические кривые измеряли на потенциостате PAR273 EG&G и регистрировали с помощью двухкоординатного самописца. Скорость развертки составляла 50 мВ/с при измерениях на золотом электроде и 10 мВ/с при измерениях на стеклоуглеродном и амальгамированном золотом электродах. Интервалы потенциалов выбирали с учетом устойчивости электродов к анодной поляризации.
Потенциодинамические кривые измеряли на потенциостате PAR273 EG&G и регистрировали с помощью двухкоординатного самописца. Скорость развертки составляла 50 мВ/с при измерениях на золотом электроде и 10 мВ/с при измерениях на стеклоуглеродном и амальгамированном золотом электродах. Интервалы потенциалов выбирали с учетом устойчивости электродов к анодной поляризации.
![]() Была сделана попытка охарактеризовать продукты восстановления [Cu2L]Cl2, получаемые в экспериментах по препаративному электролизу с разными степенями превращения.
Была сделана попытка охарактеризовать продукты восстановления [Cu2L]Cl2, получаемые в экспериментах по препаративному электролизу с разными степенями превращения.
![]() Электролиз выполняли в плоскодонной (диаметр дна около 8 см) ячейке с разделенными пространствами, в которой рабочим электродом служила донная ртуть, а вспомогательным электродом платиновая пластинка. В ходе электролиза раствор и ртуть перемешивали током аргона. Конструкция ячейки позволяла изолировать раствор после электролиза в атмосфере аргона. Восстановление проводили, используя потенциостат П-5827, при потенциале –0.6 В в 0.89, 1 и 3мМ растворах [Cu2L]Cl2 (25 мл) в 0.1 М ацетатном буферном растворе (pH 6.0). Количество пропущенного заряда рассчитывали путем интегрирования кривой зависимости тока от времени. Заряд, определенный в контрольном эксперименте по электролизу в фоновом электролите, составлял не более 5% (он, по-видимому, затрачивался на восстановление следов кислорода).
Электролиз выполняли в плоскодонной (диаметр дна около 8 см) ячейке с разделенными пространствами, в которой рабочим электродом служила донная ртуть, а вспомогательным электродом платиновая пластинка. В ходе электролиза раствор и ртуть перемешивали током аргона. Конструкция ячейки позволяла изолировать раствор после электролиза в атмосфере аргона. Восстановление проводили, используя потенциостат П-5827, при потенциале –0.6 В в 0.89, 1 и 3мМ растворах [Cu2L]Cl2 (25 мл) в 0.1 М ацетатном буферном растворе (pH 6.0). Количество пропущенного заряда рассчитывали путем интегрирования кривой зависимости тока от времени. Заряд, определенный в контрольном эксперименте по электролизу в фоновом электролите, составлял не более 5% (он, по-видимому, затрачивался на восстановление следов кислорода).
![]() Количество невосстановленного комплекса оценивали полярографически после окончания электролиза.
Количество невосстановленного комплекса оценивали полярографически после окончания электролиза.
![]() Для характеристики осадка, образовавшегося в ходе электролиза, использовали метод ПМР (DPX-300, Bruker ) с рабочей частотой 300 МГц.
Для характеристики осадка, образовавшегося в ходе электролиза, использовали метод ПМР (DPX-300, Bruker ) с рабочей частотой 300 МГц.
![]() Регистрацию спектров поглощения растворов в ультрафиолетовой и видимой областях проводили на фотометре КФК-3, который позволял измерять оптическую плотность в интервале длин волн от 360 до 800 нм. Толщина кварцевых кювет составляла 1.0 см.
Регистрацию спектров поглощения растворов в ультрафиолетовой и видимой областях проводили на фотометре КФК-3, который позволял измерять оптическую плотность в интервале длин волн от 360 до 800 нм. Толщина кварцевых кювет составляла 1.0 см.
![]() Первоначально электрохимическое поведение [Cu2L]Cl2 изучали на стеклоуглеродном электроде, позволяющем работать в достаточно широком интервале потенциалов.
Первоначально электрохимическое поведение [Cu2L]Cl2 изучали на стеклоуглеродном электроде, позволяющем работать в достаточно широком интервале потенциалов.
![]() При введении [Cu2L]Cl2 практически во всем интервале потенциалов от 0.2 до –1 В наблюдалось снижение токов в 2 раза по сравнению с фоновыми. Это позволило предположить сильную адсорбцию комплекса [Cu2L]Cl2 на поверхности стеклоуглерода.
При введении [Cu2L]Cl2 практически во всем интервале потенциалов от 0.2 до –1 В наблюдалось снижение токов в 2 раза по сравнению с фоновыми. Это позволило предположить сильную адсорбцию комплекса [Cu2L]Cl2 на поверхности стеклоуглерода.
![]() Потенциодинамические кривые в растворе [Cu2L]Cl2 при pH 6.0 на стеклоуглероде отличаются от фоновых кривых слабо выраженным размытым катодным пиком при потенциале –0.7 В, высота которого не превышает 10% от токов заряжения в данном интервале потенциалов.
Потенциодинамические кривые в растворе [Cu2L]Cl2 при pH 6.0 на стеклоуглероде отличаются от фоновых кривых слабо выраженным размытым катодным пиком при потенциале –0.7 В, высота которого не превышает 10% от токов заряжения в данном интервале потенциалов.
![]() Восстановление комплекса [Cu2L]Cl2 в ходе линейной развертки потенциала удается наблюдать на амальгамированном золотом электроде (рис.2).
Восстановление комплекса [Cu2L]Cl2 в ходе линейной развертки потенциала удается наблюдать на амальгамированном золотом электроде (рис.2).
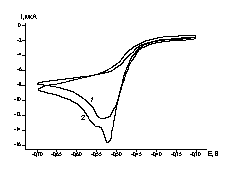 |
| Рис. 2. Циклические вольтамперограммы, измеренные на амальгамированном золотом электроде в 0.1 М (AcH + AcNa) (pH 6.0) с добавками 0.3 мМ [Cu2L]Cl2 при скорости развертки 10 мВ/с: 1 – 5, 2 - 20 цикл. |
![]() Ход потенциодинамических кривых на амальгамированном электроде чувствителен к величинам потенциалов границ циклирования, а также несколько изменяется в последовательных циклах. Если потенциал катодной границы циклирования составляет не менее –0.7 В, то в первых пяти циклах удается наблюдать кривую с одним катодным пиком тока (кривая 1 рис.2). В последующих циклах на катодном ходе в той же области потенциалов от –0.3 до –0.7 В появляется дополнительное плечо. На анодном ходе кривых в области потенциалов, отвечающих устойчивости амальгамы, не удается зарегистрировать откликов продуктов, что указывает на выраженную необратимость процесса, возможно связанную с протеканием последовательных химических стадий.
Ход потенциодинамических кривых на амальгамированном электроде чувствителен к величинам потенциалов границ циклирования, а также несколько изменяется в последовательных циклах. Если потенциал катодной границы циклирования составляет не менее –0.7 В, то в первых пяти циклах удается наблюдать кривую с одним катодным пиком тока (кривая 1 рис.2). В последующих циклах на катодном ходе в той же области потенциалов от –0.3 до –0.7 В появляется дополнительное плечо. На анодном ходе кривых в области потенциалов, отвечающих устойчивости амальгамы, не удается зарегистрировать откликов продуктов, что указывает на выраженную необратимость процесса, возможно связанную с протеканием последовательных химических стадий.
![]() При смещении катодной границы циклирования до –1.1 В на вольтамперограммах наблюдается второй широкий пик при –0.7 В, форма которого изменяется от цикла к циклу. Полученные данные указывают на существенное изменение состояния поверхности амальгамы в результате восстановления реагента. По-видимому, на поверхности аккумулируются адсорбированные или твердые продукты реакции.
При смещении катодной границы циклирования до –1.1 В на вольтамперограммах наблюдается второй широкий пик при –0.7 В, форма которого изменяется от цикла к циклу. Полученные данные указывают на существенное изменение состояния поверхности амальгамы в результате восстановления реагента. По-видимому, на поверхности аккумулируются адсорбированные или твердые продукты реакции.
![]() На рис. 3 приведены потенциодинамические кривые, полученные на золотом электроде, позволяющем расширить интервал потенциалов в сторону более положительных значений. Увеличение катодного тока по сравнению с фоновым становится заметным при потенциалах отрицательнее –0.4 В (рис. 3а), как и в случае измерений на амальгамированном золотом электроде. Площадка катодного тока начинается при потенциале –0.7 В (рис. 3б). На обратном ходе удается наблюдать пик анодного тока при 0.3 В, а при смещении катодного предела потенциала до –0.8 В появление второго анодного пика при 0.05 В. Заряд, отвечающий анодным пикам, близок к заряду, пропущенному на катодном ходе кривых, если катодный предел развертки составляет от –0.5 до –0.9 В. При дальнейшем смещении катодного предела баланс нарушается, что указывает на изменение состава продуктов. Потенциалы анодных пиков отвечают характерной области десорбции адатомов меди и окисления фазы меди (около двух монослоев). Для подтверждения этого предположения были проведены контрольные опыты в фоновом растворе с добавками 20 мМ сульфата меди. Потенциал катодной границы составлял 0 и –0.03 В, то есть отвечал фазовому осаждению меди. Пик десорбции меди на циклических вольтамперных кривых наблюдали при потенциалах 0.2 и 0.3 В. Более тонкое исследование в области образования адатомов меди при pH 6.0 осложняется образованием оксокомплексов меди. Однако, установленные в тестовых опытах, потенциалы десорбции близки к потенциалам пиков на анодном ходе кривых (рис. 3б). Это позволяет предположить, что единственным медь-содержащим продуктом восстановления при потенциалах отрицательнее –0.9 В является металлическая медь. Такое предположение означает, что восстановление происходит с переносом четырех электронов. Четырехэлектронное восстановление КРТ при одном и том же потенциале в неводных средах в литературе не описано.
На рис. 3 приведены потенциодинамические кривые, полученные на золотом электроде, позволяющем расширить интервал потенциалов в сторону более положительных значений. Увеличение катодного тока по сравнению с фоновым становится заметным при потенциалах отрицательнее –0.4 В (рис. 3а), как и в случае измерений на амальгамированном золотом электроде. Площадка катодного тока начинается при потенциале –0.7 В (рис. 3б). На обратном ходе удается наблюдать пик анодного тока при 0.3 В, а при смещении катодного предела потенциала до –0.8 В появление второго анодного пика при 0.05 В. Заряд, отвечающий анодным пикам, близок к заряду, пропущенному на катодном ходе кривых, если катодный предел развертки составляет от –0.5 до –0.9 В. При дальнейшем смещении катодного предела баланс нарушается, что указывает на изменение состава продуктов. Потенциалы анодных пиков отвечают характерной области десорбции адатомов меди и окисления фазы меди (около двух монослоев). Для подтверждения этого предположения были проведены контрольные опыты в фоновом растворе с добавками 20 мМ сульфата меди. Потенциал катодной границы составлял 0 и –0.03 В, то есть отвечал фазовому осаждению меди. Пик десорбции меди на циклических вольтамперных кривых наблюдали при потенциалах 0.2 и 0.3 В. Более тонкое исследование в области образования адатомов меди при pH 6.0 осложняется образованием оксокомплексов меди. Однако, установленные в тестовых опытах, потенциалы десорбции близки к потенциалам пиков на анодном ходе кривых (рис. 3б). Это позволяет предположить, что единственным медь-содержащим продуктом восстановления при потенциалах отрицательнее –0.9 В является металлическая медь. Такое предположение означает, что восстановление происходит с переносом четырех электронов. Четырехэлектронное восстановление КРТ при одном и том же потенциале в неводных средах в литературе не описано.
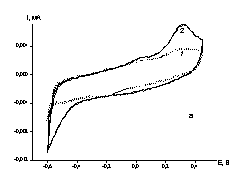 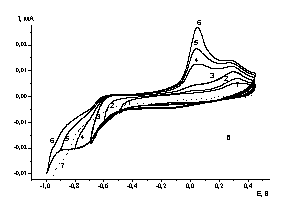 |
| Рис. 3. Циклические вольтамперограммы, измеренные на золотом электроде при скорости развертки 50 мВ/с, а) 1 - в 0.1 М (AcH + AcNa) (pH 6.0), 2- в растворе 0.1 М ацетатный буфер pH 6.0 + 0.3 мМ [Cu2L]Cl2; б) в растворе 0.1 М (AcH + AcNa) (pH 6.0) + 0.3 мМ [Cu2L]Cl2 при катодных пределах потенциалов, В: 1 - -0.5, 2 - -0.6, 3 - -0.7, 4 - -0.8, 5 - - 0.9, 6 - -1. Кривая 7 – катодный ход в растворе фона. |
![]() В случае смещения катодной границы циклирования до –1 В ток в рабочих растворах все еще остается меньшим, чем в растворе фона при тех же потенциалах (кривая 7 рис. 3б). По-видимому, имеет место существенное ингибирование процесса выделения водорода в присутствии КРТ, что косвенно указывает на адсорбцию реагента и (или) продуктов.
В случае смещения катодной границы циклирования до –1 В ток в рабочих растворах все еще остается меньшим, чем в растворе фона при тех же потенциалах (кривая 7 рис. 3б). По-видимому, имеет место существенное ингибирование процесса выделения водорода в присутствии КРТ, что косвенно указывает на адсорбцию реагента и (или) продуктов.
![]() Токи восстановлении комплекса [Cu2L]Cl2 на ртутном капельном электроде при pH 6.0 удается зафиксировать при потенциалах отрицательнее –0.4 В (кривая 1 рис.4), то есть потенциал начала восстановления Ек1 практически одинаков при измерениях на ртутном, амальгамированном и золотом электродах (таблица). На начальном участке полярограммы имеется волна с четко выраженным участком предельного диффузионного тока. При потенциалах отрицательнее –0.8 В наблюдаются еще две волны восстановления, суммарная высота которых близка к высоте первой волны.
Токи восстановлении комплекса [Cu2L]Cl2 на ртутном капельном электроде при pH 6.0 удается зафиксировать при потенциалах отрицательнее –0.4 В (кривая 1 рис.4), то есть потенциал начала восстановления Ек1 практически одинаков при измерениях на ртутном, амальгамированном и золотом электродах (таблица). На начальном участке полярограммы имеется волна с четко выраженным участком предельного диффузионного тока. При потенциалах отрицательнее –0.8 В наблюдаются еще две волны восстановления, суммарная высота которых близка к высоте первой волны.
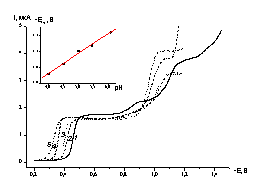 |
| Рис. 4. Полярограммы, измеренные на капающем ртутном электроде в растворах 0.3 мМ [Cu2L]Cl2 + 0.1 М (AcH + AcNa), pH: 1 - 6.0, 2 -5.45, 3 - 5.0, 4 - 4.5, 5 - 4.0. Вставка - зависимость потенциала полуволны от pH. |
![]() Токи, измеренные при потенциалах –0.6, -1 и –1.2 В (pH 6.0), пропорциональны квадратному корню из высоты ртутного столба, что подтверждает диффузионную природу всех зарегистрированных предельных токов. Предельный ток первой волны пропорционален концентрации [Cu2L]Cl2, по крайней мере до концентрации комплекса 1мМ, отклонение от линейной зависимости не превышает 5%.
Токи, измеренные при потенциалах –0.6, -1 и –1.2 В (pH 6.0), пропорциональны квадратному корню из высоты ртутного столба, что подтверждает диффузионную природу всех зарегистрированных предельных токов. Предельный ток первой волны пропорционален концентрации [Cu2L]Cl2, по крайней мере до концентрации комплекса 1мМ, отклонение от линейной зависимости не превышает 5%.
![]() Первая волна на полярограмме отвечает тому же интервалу потенциалов, в котором на золотом электроде фиксировалось образование металлической меди. По уравнению Ильковича из предельного диффузионного тока первой волны (рН 6.0) были вычислены возможные значения коэффициента диффузии D в различных предположениях о числе переносимых электронов. В области первой волны D = (8 ± 3)·10-6 cм2/с в предположении о двухэлектронном восстановлении и D = (2 ± 1)·10-6 cм2/с в предположении о четырехэлектронном процессе.
Первая волна на полярограмме отвечает тому же интервалу потенциалов, в котором на золотом электроде фиксировалось образование металлической меди. По уравнению Ильковича из предельного диффузионного тока первой волны (рН 6.0) были вычислены возможные значения коэффициента диффузии D в различных предположениях о числе переносимых электронов. В области первой волны D = (8 ± 3)·10-6 cм2/с в предположении о двухэлектронном восстановлении и D = (2 ± 1)·10-6 cм2/с в предположении о четырехэлектронном процессе.
![]() Значение коэффициента диффузии, вычисленное по данным работы [6] для комплекса, отличающегося от [Cu2L]Cl2 4-алкильным заместителем и природой противоина, составляет 4.84·10-6 cм2/с для одноэлектронного восстановления в диметилформамиде. Пересчет значения D, приведенного в работе [13], для КРТ меди на основе 2,6-диацетил- 4-метилфенола и 1,3-диминопропана, приводит к величине 8·10-7 см2/с для одноэлектронного восстановления в диметилформамиде. Близкая геометрия этих и исследуемого нами КРТ позволяет ожидать и близкие значения коэффициентов диффузии. С учетом отношения вязкоcтей диметилформамида и воды, значение коэффициента диффузии (8 ± 3)·10-6 cм2/с, полученное в воде для [Cu2L]Cl2, является, по-видимому, нереалистично высоким.
Значение коэффициента диффузии, вычисленное по данным работы [6] для комплекса, отличающегося от [Cu2L]Cl2 4-алкильным заместителем и природой противоина, составляет 4.84·10-6 cм2/с для одноэлектронного восстановления в диметилформамиде. Пересчет значения D, приведенного в работе [13], для КРТ меди на основе 2,6-диацетил- 4-метилфенола и 1,3-диминопропана, приводит к величине 8·10-7 см2/с для одноэлектронного восстановления в диметилформамиде. Близкая геометрия этих и исследуемого нами КРТ позволяет ожидать и близкие значения коэффициентов диффузии. С учетом отношения вязкоcтей диметилформамида и воды, значение коэффициента диффузии (8 ± 3)·10-6 cм2/с, полученное в воде для [Cu2L]Cl2, является, по-видимому, нереалистично высоким.
![]() Таким образом, предварительный анализ полярографических данных по крайней мере не противоречит предположению о четырехэлектронной природе восстановления.
Таким образом, предварительный анализ полярографических данных по крайней мере не противоречит предположению о четырехэлектронной природе восстановления.
![]() В исследованном интервале pH (кривые 1-5 рис.4) наблюдалось смещение обеих волн. Однако только для первой волны оно носило систематический характер (вставка к рис.4). Потенциал полуволны с ростом pH смещался в сторону более отрицательных значений на 64 ± 3 мВ /pH. Это соответствует присоединению одного протона при переносе каждого электрона.
В исследованном интервале pH (кривые 1-5 рис.4) наблюдалось смещение обеих волн. Однако только для первой волны оно носило систематический характер (вставка к рис.4). Потенциал полуволны с ростом pH смещался в сторону более отрицательных значений на 64 ± 3 мВ /pH. Это соответствует присоединению одного протона при переносе каждого электрона.
![]() Для более точного определения числа переносимых электронов была предпринята попытка препаративного потенциостатического электролиза при потенциале площадки предельного тока первой волны.
Для более точного определения числа переносимых электронов была предпринята попытка препаративного потенциостатического электролиза при потенциале площадки предельного тока первой волны.
![]() В ходе восстановления комплекса [Cu2L]Cl2 на ртутном электроде при –0.6 В наблюдаются следующие качественные изменения: окраска раствора довольно быстро (при заведомо неполном превращении) переходит от зеленой к салатовой и затем к желтой. После размыкания цепи и особенно при переливании раствора, содержащего продукты восстановления, заметно медленное выпадение осадка желтого цвета. При выдерживании раствора после электролиза на воздухе в течение нескольких дней цвет раствора изменяется от желтого к салатовому.
В ходе восстановления комплекса [Cu2L]Cl2 на ртутном электроде при –0.6 В наблюдаются следующие качественные изменения: окраска раствора довольно быстро (при заведомо неполном превращении) переходит от зеленой к салатовой и затем к желтой. После размыкания цепи и особенно при переливании раствора, содержащего продукты восстановления, заметно медленное выпадение осадка желтого цвета. При выдерживании раствора после электролиза на воздухе в течение нескольких дней цвет раствора изменяется от желтого к салатовому.
![]() Для определения степени превращения комплекса [Cu2L]Cl2 в результате восстановления привлекали несколько независимых методов.
Для определения степени превращения комплекса [Cu2L]Cl2 в результате восстановления привлекали несколько независимых методов.
![]() Сведений о спектрах поглощения водных растворов комплекса [Cu2L]Cl2 нами не обнаружено. Измеренный спектр поглощения [Cu2L]Cl2 в ацетатном буферном растворе (рис.5) демонстрирует два характеристических участка - край широкой полосы в области 300-450 нм и широкую полосу с максимумом около 600 нм (коэффициент экстинкции 70 л·моль-1·см-1). Подобный характер поглощения близок к описанному в литературе для водных растворов биядерного КРТ меди с лигандом на основе 4-метил-2,6-диформилфенола и 1,3-диаминопропана, отличающего от [Cu2L]Cl2 заместителем в четвертом положении фенольного кольца [3]. Согласно работе [3], полоса в области 300-450 нм обусловлена поглощением лиганда, а полоса с максимумом около 600 нм соответствует d-d переходу меди в комплексе [Cu2L]Cl2.
Сведений о спектрах поглощения водных растворов комплекса [Cu2L]Cl2 нами не обнаружено. Измеренный спектр поглощения [Cu2L]Cl2 в ацетатном буферном растворе (рис.5) демонстрирует два характеристических участка - край широкой полосы в области 300-450 нм и широкую полосу с максимумом около 600 нм (коэффициент экстинкции 70 л·моль-1·см-1). Подобный характер поглощения близок к описанному в литературе для водных растворов биядерного КРТ меди с лигандом на основе 4-метил-2,6-диформилфенола и 1,3-диаминопропана, отличающего от [Cu2L]Cl2 заместителем в четвертом положении фенольного кольца [3]. Согласно работе [3], полоса в области 300-450 нм обусловлена поглощением лиганда, а полоса с максимумом около 600 нм соответствует d-d переходу меди в комплексе [Cu2L]Cl2.
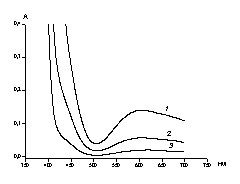 |
| Рис. 5. Спектры поглощения в растворе 0.1 М (AcH + AcNa) (pH 6.0) + [Cu2L]Cl2 при концентрациях, мМ: 1 - 2 ,2 – 0.8 ,3 - 0.3. |
![]() Наличие осадка существенно затрудняло спектрофотометрические исследования продуктов электролиза. После отфильтровывания или отделения осадка (эти операции выполнялись на воздухе) в спектрах фиксировалось сильное фоновое поглощение продукта в интервале от 500 до 700 нм, на фоне которого определение концентрации исходного реагента по его характеристической полосе не представлялось возможным.
Наличие осадка существенно затрудняло спектрофотометрические исследования продуктов электролиза. После отфильтровывания или отделения осадка (эти операции выполнялись на воздухе) в спектрах фиксировалось сильное фоновое поглощение продукта в интервале от 500 до 700 нм, на фоне которого определение концентрации исходного реагента по его характеристической полосе не представлялось возможным.
![]() Осадок, образовавшийся в результате эксперимента по восстановлению комплекса [Cu2L]Cl2, был отделен декантацией, растворен в спирте, остаток после отгонки спирта высушен и проанализирован методом 1Н ЯМР. Отсутствие в спектре уширенных сигналов, вызванных присутствием парамагнитных примесей медного комплекса, позволяет сделать вывод о полном протекании деметаллирования исходного [Cu2L]Cl2 в ходе электролиза. В спектре, снятом в CDCl3, присутствуют сигналы протонов трех трет-бутильных групп (1.26, 1.32, 1.36 м.д.), трех ароматических групп (7.99, 7.92, 7.62 м.д.), двух формильных групп (10.53 и 10.26 м.д.) и сигналы при 8.5 м.д. (N=CH), 3.8 м.д. (CH2CH2CH2) и 2.2 м.д. (CH2CH2CH2). Это может свидетельствовать о наличии по крайней мере трех продуктов: исходного лиганда [19], продукта частичного гидролиза 1.3-бис[(2-гидрокси-5-трет-бутил-3-формилфенил)метилен]аминопропана, образующегося в результате элиминирования одной молекуля диаминопропана, и продукта полного гидролиза с элиминированием двух молекул диаминопропана - 4-трет-бутил-2.6-диформилфенола [20]. Таким образом, спектр ПМР осадка, образующегося после восстановления [Cu2L]Cl2, свидетельствует по крайней мере о частичной ступенчатой деструкции макроциклического лиганда в процессе восстановления.
Осадок, образовавшийся в результате эксперимента по восстановлению комплекса [Cu2L]Cl2, был отделен декантацией, растворен в спирте, остаток после отгонки спирта высушен и проанализирован методом 1Н ЯМР. Отсутствие в спектре уширенных сигналов, вызванных присутствием парамагнитных примесей медного комплекса, позволяет сделать вывод о полном протекании деметаллирования исходного [Cu2L]Cl2 в ходе электролиза. В спектре, снятом в CDCl3, присутствуют сигналы протонов трех трет-бутильных групп (1.26, 1.32, 1.36 м.д.), трех ароматических групп (7.99, 7.92, 7.62 м.д.), двух формильных групп (10.53 и 10.26 м.д.) и сигналы при 8.5 м.д. (N=CH), 3.8 м.д. (CH2CH2CH2) и 2.2 м.д. (CH2CH2CH2). Это может свидетельствовать о наличии по крайней мере трех продуктов: исходного лиганда [19], продукта частичного гидролиза 1.3-бис[(2-гидрокси-5-трет-бутил-3-формилфенил)метилен]аминопропана, образующегося в результате элиминирования одной молекуля диаминопропана, и продукта полного гидролиза с элиминированием двух молекул диаминопропана - 4-трет-бутил-2.6-диформилфенола [20]. Таким образом, спектр ПМР осадка, образующегося после восстановления [Cu2L]Cl2, свидетельствует по крайней мере о частичной ступенчатой деструкции макроциклического лиганда в процессе восстановления.
![]() Наиболее информативным оказалось определение остаточного количества реагента в смеси после электролиза полярографическим методом по величине предельного диффузионного тока первой волны. В принципе, нельзя исключить, что какой-либо из продуктов восстановления образовавшийся в ходе препаративного электролиза, дает, наряду с остаточным реагентом, вклад в первую волну восстановления. Это, однако, означало бы существование различных соединений с очень близкими потенциалами полуволн, что крайне маловероятно. Для проверки предположения об образовании 4-трет-бутил-2,6-диформилфенола как возможного продукта деструкции восстановленного [Cu2L]Cl2 были проведены контрольные полярографические измерения в 0.1 М ацетатном буферном растворе (pH 6.0) с добавлением 4-трет-бутил-2,6-диформилфенола. Начало волны восстановления на полярограмме наблюдается при потенциале –1 В. Это означает, что возможное присутствие 4-трет-бутил-2,6-диформилфенола в смеси после электролиза не вносило бы вклад в величину предельного тока [Cu2L]Cl2 на первой волне.
Наиболее информативным оказалось определение остаточного количества реагента в смеси после электролиза полярографическим методом по величине предельного диффузионного тока первой волны. В принципе, нельзя исключить, что какой-либо из продуктов восстановления образовавшийся в ходе препаративного электролиза, дает, наряду с остаточным реагентом, вклад в первую волну восстановления. Это, однако, означало бы существование различных соединений с очень близкими потенциалами полуволн, что крайне маловероятно. Для проверки предположения об образовании 4-трет-бутил-2,6-диформилфенола как возможного продукта деструкции восстановленного [Cu2L]Cl2 были проведены контрольные полярографические измерения в 0.1 М ацетатном буферном растворе (pH 6.0) с добавлением 4-трет-бутил-2,6-диформилфенола. Начало волны восстановления на полярограмме наблюдается при потенциале –1 В. Это означает, что возможное присутствие 4-трет-бутил-2,6-диформилфенола в смеси после электролиза не вносило бы вклад в величину предельного тока [Cu2L]Cl2 на первой волне.
![]() Полярограммы растворов, полученных в результате восстановления [Cu2L]Cl2 с различными степенями превращения, на начальном участке содержат лишь волну восстановления [Cu2L]Cl2. Величина предельного диффузионного тока первой волны практически не изменяется при измерении полярограммы сразу после восстановления и через несколько дней хранения раствора на воздухе или после отфильтровывания осадка. Типичная полярограмма раствора, подвергнутого электролизу, представлена на рис. 6 (кривые 2 и 3). Это означает, что окисление кислородом воздуха не приводит к образованию каких-либо веществ, электрохимически активных в той же области потенциалов, то есть какие-либо погрешности, связанные с несовершенством процедуры переноса раствора после электролиза в полярографическую ячейку, исключены.
Полярограммы растворов, полученных в результате восстановления [Cu2L]Cl2 с различными степенями превращения, на начальном участке содержат лишь волну восстановления [Cu2L]Cl2. Величина предельного диффузионного тока первой волны практически не изменяется при измерении полярограммы сразу после восстановления и через несколько дней хранения раствора на воздухе или после отфильтровывания осадка. Типичная полярограмма раствора, подвергнутого электролизу, представлена на рис. 6 (кривые 2 и 3). Это означает, что окисление кислородом воздуха не приводит к образованию каких-либо веществ, электрохимически активных в той же области потенциалов, то есть какие-либо погрешности, связанные с несовершенством процедуры переноса раствора после электролиза в полярографическую ячейку, исключены.
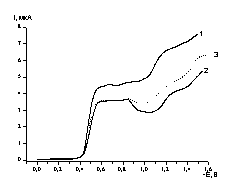 |
| Рис. 6. Полярографические кривые на капающем ртутном электроде: кривая 1 – исходный раствор 1 мМ [Cu2L]Cl2 + 0.1 М (AcH + AcNa) (pH 6.0), 2 – раствор непосредственно после электролиза, 3 – раствор после электролиза, отфильтрованный от выпавшего осадка. |
![]() В нескольких экспериментах на полярограммах наблюдались спады тока в интервале потенциалов от –0.8 до –1.2 В (рис. 6), однако величина предельного тока на первой волне оставалась неизменной. Полярографическое определение остаточного содержания комплекса оказалось возможным благодаря необратимому характеру восстановления. Принятые при переносе раствора предосторожности (продувка раствора после электролиза аргоном во вспомогательном сосуде) в данном случае были, по-видимому, не столь существенны.
В нескольких экспериментах на полярограммах наблюдались спады тока в интервале потенциалов от –0.8 до –1.2 В (рис. 6), однако величина предельного тока на первой волне оставалась неизменной. Полярографическое определение остаточного содержания комплекса оказалось возможным благодаря необратимому характеру восстановления. Принятые при переносе раствора предосторожности (продувка раствора после электролиза аргоном во вспомогательном сосуде) в данном случае были, по-видимому, не столь существенны.
![]() На рис.7 приведена зависимость определенной полярографически степени превращения комплекса [Cu2L]Cl2 (точки) от пропущенного в ходе электролиза заряда. Прямые 1 и 2 отвечают теоретическим зависимостям, рассчитанным в предположениях о четырех- (кривая 1) и двух- (кривая 2) электронном восстановлении соответственно. Из рис.7 видно, что экспериментально полученные значения степеней превращения близки к прямой 1. Поскольку в пользу предположения о четырехэлектронном восстановлении свидетельствуют также более близкое к литературным данным значение коэффициента диффузии [Cu2L]Cl2 (D = (2 ± 1)·10-6 cм2/с) и кулонометрические данные, полученные на золотом электроде, следует признать предположение о четырехэлектронной природе первой волны вполне реалистичным.
На рис.7 приведена зависимость определенной полярографически степени превращения комплекса [Cu2L]Cl2 (точки) от пропущенного в ходе электролиза заряда. Прямые 1 и 2 отвечают теоретическим зависимостям, рассчитанным в предположениях о четырех- (кривая 1) и двух- (кривая 2) электронном восстановлении соответственно. Из рис.7 видно, что экспериментально полученные значения степеней превращения близки к прямой 1. Поскольку в пользу предположения о четырехэлектронном восстановлении свидетельствуют также более близкое к литературным данным значение коэффициента диффузии [Cu2L]Cl2 (D = (2 ± 1)·10-6 cм2/с) и кулонометрические данные, полученные на золотом электроде, следует признать предположение о четырехэлектронной природе первой волны вполне реалистичным.
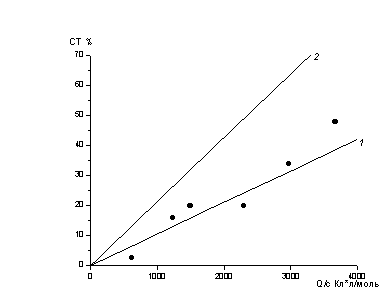 |
| Рис. 7. Зависимость теоретической и экспериментальной степени превращения [Cu2L]Cl2 (СТ%) от относительного количества пропущенного заряда (Q/c, где Q-заряд, Кл, с - концентрация [Cu2L]Cl2, М): 1 –теоретическая кривая для четырехэлектронного восстановления, 2 - для двухэлектронного, точки - экспериментальные данные. |
![]() Предполагаемая общая схема восстановления изучаемого комплекса [Cu2L]Cl2. включает восстановление обоих ионов меди до металлической меди, сопровождающееся деструкцией [Cu2L]Cl2 и образованием свободного лиганда, который, вероятно, претерпевает дальнейшие химические превращения, например, гидролиз.
Предполагаемая общая схема восстановления изучаемого комплекса [Cu2L]Cl2. включает восстановление обоих ионов меди до металлической меди, сопровождающееся деструкцией [Cu2L]Cl2 и образованием свободного лиганда, который, вероятно, претерпевает дальнейшие химические превращения, например, гидролиз.
| [Cu(II)Cu(II)L]2+ + 4e- + 4H+ --> 2Cu + (H4L2+), |
где [Cu(II)Cu(II)L]2+ –исходный комплекс, H4L2+ –макроциклический лиганд.
![]() Нельзя исключить возможность промежуточного образования комплекса, в котором оба иона меди находятся в состоянии Cu(I), и последующего восстановления лиганда, которое сопровождается протонированием и приводит к разрушению комплекса. Такой ход восстановления не противоречил бы экспериментальным данным при обнаружении восстановленного лиганда или продуктов его деструкции в смеси после электролиза. Также оказывается возможным диспропорционирование образующегося комплекса [Cu(I)Cu(I)L] до металлической меди и меди в состоянии Cu(II).
Нельзя исключить возможность промежуточного образования комплекса, в котором оба иона меди находятся в состоянии Cu(I), и последующего восстановления лиганда, которое сопровождается протонированием и приводит к разрушению комплекса. Такой ход восстановления не противоречил бы экспериментальным данным при обнаружении восстановленного лиганда или продуктов его деструкции в смеси после электролиза. Также оказывается возможным диспропорционирование образующегося комплекса [Cu(I)Cu(I)L] до металлической меди и меди в состоянии Cu(II).
![]() Разумеется, приведенная выше схема представляет собой сложную реакцию, которая включает отдельные элементарные превращения.
Разумеется, приведенная выше схема представляет собой сложную реакцию, которая включает отдельные элементарные превращения.
![]() Для КРТ [Cu2L]Cl2, электрохимическое поведение которого, как и его разнообразных аналогов, ранее не изучалось в водной среде, выявлены следующие особенности электровосстановления. Комплекс [Cu2L]Cl2 сильно адсорбируется на поверхности стеклоуглерода, что затрудняет изучение восстановления на этом электродном материале. На ртутном капающем электроде [Cu2L]Cl2 потенциал полуволны составляет –0.46 – 0.06/pH В относительно нас. к. э. Последующее восстановление скорее всего осложнено химическими стадиями. Потенциалы начала восстановления [Cu2L]Cl2 в водной среде при pH 6.0 на всех исследованных электродных материалов совпадают. Зафиксировано в опытах по препаративному электролизу образование металлической меди.
Для КРТ [Cu2L]Cl2, электрохимическое поведение которого, как и его разнообразных аналогов, ранее не изучалось в водной среде, выявлены следующие особенности электровосстановления. Комплекс [Cu2L]Cl2 сильно адсорбируется на поверхности стеклоуглерода, что затрудняет изучение восстановления на этом электродном материале. На ртутном капающем электроде [Cu2L]Cl2 потенциал полуволны составляет –0.46 – 0.06/pH В относительно нас. к. э. Последующее восстановление скорее всего осложнено химическими стадиями. Потенциалы начала восстановления [Cu2L]Cl2 в водной среде при pH 6.0 на всех исследованных электродных материалов совпадают. Зафиксировано в опытах по препаративному электролизу образование металлической меди.
![]() Величины коэффициентов диффузии [Cu2L]Cl2, вычисленные из предельного диффузионного тока, и данные экспериментов по препаративному электролизу являются серьезными аргументами в пользу предположения о четырехэлектронном процессе при потенциалах первой волны. Большой интерес представляют дальнейшие эксперименты по разделению этой волны в смешанных водно-органических растворителях и согласование разностей потенциалов последовательных превращений в средах с различной диэлектрической проницаемостью. Эти данные могут стать чрезвычайно полезными для экспериментальной проверки теории [11].
Величины коэффициентов диффузии [Cu2L]Cl2, вычисленные из предельного диффузионного тока, и данные экспериментов по препаративному электролизу являются серьезными аргументами в пользу предположения о четырехэлектронном процессе при потенциалах первой волны. Большой интерес представляют дальнейшие эксперименты по разделению этой волны в смешанных водно-органических растворителях и согласование разностей потенциалов последовательных превращений в средах с различной диэлектрической проницаемостью. Эти данные могут стать чрезвычайно полезными для экспериментальной проверки теории [11].
![]() Работа выполнена при поддержке РФФИ (проекты 02-03-33321а и 02-03-32101) и Совета по грантам Президента Российской Федерации для поддержки научных школ (НШ – 2089.2003.3).
Работа выполнена при поддержке РФФИ (проекты 02-03-33321а и 02-03-32101) и Совета по грантам Президента Российской Федерации для поддержки научных школ (НШ – 2089.2003.3).
| ||||||||
| # уширенный, плохо воспроизводимый пик ## потенциал начала площадки катодного тока |