![]() Дана общая характеристика кинетики электровосстановления церий-декавольфрамат-аниона на ртути, поликристаллическом золоте и базисной грани пирографита в ацетатных буферных растворах (рН 3.5 - 6.0). На основании анализа УФ-спектров поглощения оценено соотношение двух форм реагента, различающихся степенью протонирования, в исследованных растворах. Показано, что при отрицательных зарядах ртутного электрода восстановление центрального иона Се(IV) протекает как внешнесферный процесс с классическими проявлениями пси-прим эффекта (наблюдается минимум тока на поляризационных кривых, глубина которого увеличивается при разбавлении электролита фона и увеличении отрицательного заряда реагента). При положительных зарядах поверхности выявлен рост тока при увеличении концентрации электролита фона, интерпретированный в предположении о прочной адсорбции реагента и его соадсорбции с катионами. В случае золотого электрода обнаружены выраженные эффекты прочной хемосорбции. Показано, что адсорбционные осложнения на пирографите проявляются тем сильнее, чем выше степень неидеальности поверхности. При низкой концентрации атомарных ступеней (по-видимому, в условиях малого заполнения поверхности пирографита адсорбированным реагентом) удается наблюдать квазиобратимый перенос электрона в системе Се(IV/III), скорость которого увеличивается с ростом рН.
Дана общая характеристика кинетики электровосстановления церий-декавольфрамат-аниона на ртути, поликристаллическом золоте и базисной грани пирографита в ацетатных буферных растворах (рН 3.5 - 6.0). На основании анализа УФ-спектров поглощения оценено соотношение двух форм реагента, различающихся степенью протонирования, в исследованных растворах. Показано, что при отрицательных зарядах ртутного электрода восстановление центрального иона Се(IV) протекает как внешнесферный процесс с классическими проявлениями пси-прим эффекта (наблюдается минимум тока на поляризационных кривых, глубина которого увеличивается при разбавлении электролита фона и увеличении отрицательного заряда реагента). При положительных зарядах поверхности выявлен рост тока при увеличении концентрации электролита фона, интерпретированный в предположении о прочной адсорбции реагента и его соадсорбции с катионами. В случае золотого электрода обнаружены выраженные эффекты прочной хемосорбции. Показано, что адсорбционные осложнения на пирографите проявляются тем сильнее, чем выше степень неидеальности поверхности. При низкой концентрации атомарных ступеней (по-видимому, в условиях малого заполнения поверхности пирографита адсорбированным реагентом) удается наблюдать квазиобратимый перенос электрона в системе Се(IV/III), скорость которого увеличивается с ростом рН.
![]() Ключевые слова: церий(IV)-декавольфрамат, ртутный капающий электрод, пирографит, поликристаллическое золото
Ключевые слова: церий(IV)-декавольфрамат, ртутный капающий электрод, пирографит, поликристаллическое золото
![]() Гетерополианионы (ГПА) с центральными атомами в высоких степенях окисления можно рассматривать как модельные системы для исследования процессов дальнего переноса электрона [1]. Действительно, если центральный ион восстанавливается при менее отрицательных потенциалах, чем оксометаллатные лиганды, то вне зависимости от локализации реагента вблизи межфазной границы расстояние переноса оказывается не меньше (а в общем случае больше), чем характерный размер оксометаллатного лиганда, т.е. превышает 0.5 нм. В отсутствие специфической адсорбции (при локализации реагента в диффузной части двойного электрического слоя) расстояние переноса электрона к центральному иону составляет около 1 нм и близко к характерным расстояниям переноса в металлопротеинах и других сложных молекулах, которые обычно рассматриваются как альтернативные модельные системы. Очевидными преимуществами обсуждаемых полиоксометаллатных реагентов являются экстремально высокие константы устойчивости, относительная простота геометрии и высокая симметрия частиц, а также инертность лиганда в отношении электронного сопряжения, что позволяет рассчитывать на достаточно простой механизм элементарного акта (отсутствие мостиковых, резонансных и других осложняющих явлений).
Гетерополианионы (ГПА) с центральными атомами в высоких степенях окисления можно рассматривать как модельные системы для исследования процессов дальнего переноса электрона [1]. Действительно, если центральный ион восстанавливается при менее отрицательных потенциалах, чем оксометаллатные лиганды, то вне зависимости от локализации реагента вблизи межфазной границы расстояние переноса оказывается не меньше (а в общем случае больше), чем характерный размер оксометаллатного лиганда, т.е. превышает 0.5 нм. В отсутствие специфической адсорбции (при локализации реагента в диффузной части двойного электрического слоя) расстояние переноса электрона к центральному иону составляет около 1 нм и близко к характерным расстояниям переноса в металлопротеинах и других сложных молекулах, которые обычно рассматриваются как альтернативные модельные системы. Очевидными преимуществами обсуждаемых полиоксометаллатных реагентов являются экстремально высокие константы устойчивости, относительная простота геометрии и высокая симметрия частиц, а также инертность лиганда в отношении электронного сопряжения, что позволяет рассчитывать на достаточно простой механизм элементарного акта (отсутствие мостиковых, резонансных и других осложняющих явлений).
![]() Исследованные ранее немногочисленные ГПА обсуждаемого типа не вполне удовлетворяют требованиям к модельным реагентам для сформулированной выше задачи. Комплексы различных переходных металлов (Mn, Co, Ni и др.) с полимолибдатными лигандами позволяют реализовать восстановление центрального иона в отсутствие параллельного восстановления лиганда и(или) окисления материала электрода лишь в узком интервале потенциалов. Более подходящими для обсуждаемой задачи представляются гетерополивольфраматы, для которых восстановление лигандов становится возможным лишь при потенциалах на сотни милливольт более отрицательных по сравнению с равновесными потенциалами редокспереходов гетероатомов переходных металлов. До сих пор кинетика восстановления центрального иона в поливольфрамате была исследована лишь для Na2K6[MnW6O24] [1]. Для этого реагента имели место специфические осложнения - двухэлектронное восстановление Mn(IV) в узком интервале потенциалов и деструкция образующегося комплекса.
Исследованные ранее немногочисленные ГПА обсуждаемого типа не вполне удовлетворяют требованиям к модельным реагентам для сформулированной выше задачи. Комплексы различных переходных металлов (Mn, Co, Ni и др.) с полимолибдатными лигандами позволяют реализовать восстановление центрального иона в отсутствие параллельного восстановления лиганда и(или) окисления материала электрода лишь в узком интервале потенциалов. Более подходящими для обсуждаемой задачи представляются гетерополивольфраматы, для которых восстановление лигандов становится возможным лишь при потенциалах на сотни милливольт более отрицательных по сравнению с равновесными потенциалами редокспереходов гетероатомов переходных металлов. До сих пор кинетика восстановления центрального иона в поливольфрамате была исследована лишь для Na2K6[MnW6O24] [1]. Для этого реагента имели место специфические осложнения - двухэлектронное восстановление Mn(IV) в узком интервале потенциалов и деструкция образующегося комплекса.
![]() Представляется более перспективным аналогичное исследование в растворах солей аниона CeW10O368- (CeW10) [2-4]. Для этого гетерополивольфрамата на основе Ce(IV) можно ожидать одноэлектронного восстановления с образованием устойчивого продукта - изоструктурного комплекса Се(III) [4-6]. Согласно рентгеноструктурным данным [3], восьмикоординированный ион церия в декавольфрамате окружен двумя фрагментами W5O18, образующими вокруг него квадратную антипризму (рис. 1) (Анион СеW10 является структурным аналогом изополивольфрамат-иона W10O324-, который также образован из двух фрагментов W5O18 с обобществлением четырех атомов кислорода.), полная симметрия D4d. Методами колебательной спектроскопии [6] и ЯМР [7] показано, что в растворах продемонстрированное на рис. 1 строение анионов СеW10 сохраняется. Спектры поглощения растворов Се(IV)W10 обнаруживают две слабо выраженные полосы в ближней УФ-области, для восстановленной формы характеристического поглощения не выявлено [8].
Представляется более перспективным аналогичное исследование в растворах солей аниона CeW10O368- (CeW10) [2-4]. Для этого гетерополивольфрамата на основе Ce(IV) можно ожидать одноэлектронного восстановления с образованием устойчивого продукта - изоструктурного комплекса Се(III) [4-6]. Согласно рентгеноструктурным данным [3], восьмикоординированный ион церия в декавольфрамате окружен двумя фрагментами W5O18, образующими вокруг него квадратную антипризму (рис. 1) (Анион СеW10 является структурным аналогом изополивольфрамат-иона W10O324-, который также образован из двух фрагментов W5O18 с обобществлением четырех атомов кислорода.), полная симметрия D4d. Методами колебательной спектроскопии [6] и ЯМР [7] показано, что в растворах продемонстрированное на рис. 1 строение анионов СеW10 сохраняется. Спектры поглощения растворов Се(IV)W10 обнаруживают две слабо выраженные полосы в ближней УФ-области, для восстановленной формы характеристического поглощения не выявлено [8].
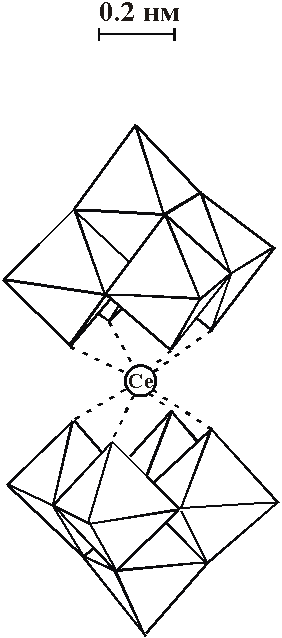 |
| Рис. 1. Схематическое изображение строения аниона [CeW10O36]8- по данным [3]. Октаэдры изображают фрагменты лиганда состава WO6. |
![]() В условиях внешнесферного переноса электрона предполагаемое расстояние переноса к СеW10 в планарной ориентации составит около 0.8 нм, в вертикальной - более 1 нм. "Ближний" перенос электрона окажется возможным только в случае существенного проникновения реагента в плотный слой, т.е. в условиях прочной хемосорбции.
В условиях внешнесферного переноса электрона предполагаемое расстояние переноса к СеW10 в планарной ориентации составит около 0.8 нм, в вертикальной - более 1 нм. "Ближний" перенос электрона окажется возможным только в случае существенного проникновения реагента в плотный слой, т.е. в условиях прочной хемосорбции.
![]() В литературе отсутствуют сведения о кинетике электровосстановления CeW10, а данные о гомогенных окислительно-восстановительных реакциях крайне отрывочны. В [2,5] приводится стандартный электродный потенциал пары Ce(IV/III) для этого комплекса, определенный методом потенциометрического титрования K4Fe(CN)6 в нейтральном растворе, он составляет +1.1 В (н.в.э.). В [9] описано окисление декавольфрамата Ce(III) озоном с образованием декавольфраматного комплекса Ce(IV). Этот результат подтверждает устойчивость координационного окружения при реокс-превращениях центрального иона церия. Таким образом, для обсуждаемого реагента, в отличие от гексавольфрамата Mn(IV), можно осуществить перенос электрона, не осложненный последующей деструкцией продукта восстановления. Настоящая работа посвящена выяснению наиболее общих закономерностей электрохимического восстановления CeW10: установлению зависимостей скорости процесса от потенциала, природы материала электрода и состава раствора.
В литературе отсутствуют сведения о кинетике электровосстановления CeW10, а данные о гомогенных окислительно-восстановительных реакциях крайне отрывочны. В [2,5] приводится стандартный электродный потенциал пары Ce(IV/III) для этого комплекса, определенный методом потенциометрического титрования K4Fe(CN)6 в нейтральном растворе, он составляет +1.1 В (н.в.э.). В [9] описано окисление декавольфрамата Ce(III) озоном с образованием декавольфраматного комплекса Ce(IV). Этот результат подтверждает устойчивость координационного окружения при реокс-превращениях центрального иона церия. Таким образом, для обсуждаемого реагента, в отличие от гексавольфрамата Mn(IV), можно осуществить перенос электрона, не осложненный последующей деструкцией продукта восстановления. Настоящая работа посвящена выяснению наиболее общих закономерностей электрохимического восстановления CeW10: установлению зависимостей скорости процесса от потенциала, природы материала электрода и состава раствора.
![]() Синтез CeW10 проводился по модифицированной методике [10] (Ce(NO3)3*6H2O заменяли на (NH4)2Ce(NO3)6). Полученное кристаллическое вещество характеризовали методом рентгеновской дифрактометрии, фазу Na6H2[CeW10O36]*30H2O идентифицировали по базе данных [3]. Отражения посторонних фаз в дифрактограмме отсутствовали. Методом термогравиметрии было определено количество гидратной воды, что позволило независимо подтвердить указанный выше состав.
Синтез CeW10 проводился по модифицированной методике [10] (Ce(NO3)3*6H2O заменяли на (NH4)2Ce(NO3)6). Полученное кристаллическое вещество характеризовали методом рентгеновской дифрактометрии, фазу Na6H2[CeW10O36]*30H2O идентифицировали по базе данных [3]. Отражения посторонних фаз в дифрактограмме отсутствовали. Методом термогравиметрии было определено количество гидратной воды, что позволило независимо подтвердить указанный выше состав.
![]() Растворы для электрохимических исследований готовили на воде, очищенной на установке Millipore. В качестве электролита фона использовали 0.01 - 0.1 М ацетатные буферные растворы, которые готовили из ледяной CH3COOH "хч" и NaOH "хч". Перед измерениями растворы деаэрировали водородом или аргоном, очищенными от следов кислорода на палладиевом или медь-хромовом катализаторах, соответственно.
Растворы для электрохимических исследований готовили на воде, очищенной на установке Millipore. В качестве электролита фона использовали 0.01 - 0.1 М ацетатные буферные растворы, которые готовили из ледяной CH3COOH "хч" и NaOH "хч". Перед измерениями растворы деаэрировали водородом или аргоном, очищенными от следов кислорода на палладиевом или медь-хромовом катализаторах, соответственно.
![]() Спектры поглощения регистрировали на спектрофотометре Unicam SP1800 в кварцевых кюветах толщиной 1 см относительно буферных растворов сравнения.
Спектры поглощения регистрировали на спектрофотометре Unicam SP1800 в кварцевых кюветах толщиной 1 см относительно буферных растворов сравнения.
![]() Полярограммы регистрировали на ртутном капающем электроде (период капания при разомкнутой цепи в фоновых растворах t = 9 - 10 c, скорость вытекания ртути m = 1.02 мг/с) в стеклянной трехэлектродной ячейке. В некоторых опытах варьировали высоту ртутного столба h в интервале 40 - 80 см. В качестве поликристаллического золотого электрода использовали золотую проволоку (99.999 %). Площадь видимой поверхности составляла 0.3 см2. Перед опытами Au электрод протравливали в течение 15 с в горячей царской водке, после чего тщательно промывали очищенной водой. В опытах с высокоориентированным пирографитом использовалась ячейка прижимного типа (электрод на дне ячейки) с малым объемом раствора. В этих условиях длительные измерения были невозможны, а точность измерения потенциала не превышала ±0.02 В. При работе с электродами из пирографита в конфигурации обычной трехэлектродной ячейки с разделенными пространствами изолировали торцевые поверхности пластин силиконовым герметиком, а плоскую рабочую поверхность (площадью 0.3 см2) обновляли непосредственно перед опытами путем скола, используя скотч [11]. Для оценки плотности ступеней на поверхности пирографита привлекали метод сканирующей туннельной микроскопии (установка LitScan-2, см. методику в [11]).
Полярограммы регистрировали на ртутном капающем электроде (период капания при разомкнутой цепи в фоновых растворах t = 9 - 10 c, скорость вытекания ртути m = 1.02 мг/с) в стеклянной трехэлектродной ячейке. В некоторых опытах варьировали высоту ртутного столба h в интервале 40 - 80 см. В качестве поликристаллического золотого электрода использовали золотую проволоку (99.999 %). Площадь видимой поверхности составляла 0.3 см2. Перед опытами Au электрод протравливали в течение 15 с в горячей царской водке, после чего тщательно промывали очищенной водой. В опытах с высокоориентированным пирографитом использовалась ячейка прижимного типа (электрод на дне ячейки) с малым объемом раствора. В этих условиях длительные измерения были невозможны, а точность измерения потенциала не превышала ±0.02 В. При работе с электродами из пирографита в конфигурации обычной трехэлектродной ячейки с разделенными пространствами изолировали торцевые поверхности пластин силиконовым герметиком, а плоскую рабочую поверхность (площадью 0.3 см2) обновляли непосредственно перед опытами путем скола, используя скотч [11]. Для оценки плотности ступеней на поверхности пирографита привлекали метод сканирующей туннельной микроскопии (установка LitScan-2, см. методику в [11]).
![]() Потенциодинамические измерения проводили при скорости развертки 0.05 В/с с помощью потенциостата/гальваностата EG&G PAR 173, снабженного универсальным программатором EG&G PAR 175. Для регистрации данных использовали двухкоординатный самописец XYRecorder A3.
Потенциодинамические измерения проводили при скорости развертки 0.05 В/с с помощью потенциостата/гальваностата EG&G PAR 173, снабженного универсальным программатором EG&G PAR 175. Для регистрации данных использовали двухкоординатный самописец XYRecorder A3.
![]() Все значения потенциала измерялись и приводятся ниже относительно насыщенного каломельного электрода.
Все значения потенциала измерялись и приводятся ниже относительно насыщенного каломельного электрода.
![]() С целью изучения кинетики восстановления центрального иона церия в анионе CeW10 были проведены полярографические исследования растворов натриевой соли реагента в ацетатном буферном растворе с постоянной концентрацией гетерополианиона и различной концентрацией электролита фона. Необходимость использования буферного раствора была вызвана тем, что некоторые соединения Ce(IV) при восстановлении протонируются. В настоящем разделе важнейшие особенности исследуемой реакции рассмотрены на примере растворов с рН 5.5.
С целью изучения кинетики восстановления центрального иона церия в анионе CeW10 были проведены полярографические исследования растворов натриевой соли реагента в ацетатном буферном растворе с постоянной концентрацией гетерополианиона и различной концентрацией электролита фона. Необходимость использования буферного раствора была вызвана тем, что некоторые соединения Ce(IV) при восстановлении протонируются. В настоящем разделе важнейшие особенности исследуемой реакции рассмотрены на примере растворов с рН 5.5.
![]() На полярограммах, полученных в достаточно разбавленных фоновых растворах (например, кривые 1, 2 рис. 2), можно выделить четыре характерных участка.
На полярограммах, полученных в достаточно разбавленных фоновых растворах (например, кривые 1, 2 рис. 2), можно выделить четыре характерных участка.
![]() (A) Участок роста тока при Е > -0.3 В. Отметим, что поправка на ток заряжения не очень точна, так как измеряемый ток в начале этого участка сравним с фоновым. Поскольку в данном случае нельзя исключить возможности параллельного окисления ртути, то трудно определить потенциал, при котором ток достигает нулевого значения и однозначно обсуждать его как потенциал начала восстановления исследуемого аниона. В то же время, можно вполне определенно утверждать, что восстановление CeW10 протекает только при потенциалах на ~1 В более отрицательных, чем стандартный потенциал, приведенный в [2,5]. Если данные [5] рассматривать как надежные (Некоторые сомнения вызывает методика титрования [5]: поскольку октациановольфрамат является более прочным комплексом, чем гексацианоферрат, в ходе титрования не исключена деструкция гетерополисоединения с образованием аквакомплексов Се(IV) и, соответственно, смещение плато на титриметрической кривой в сторону более высоких потенциалов.), это различие можно интерпретировать только в предположении о чрезвычайно низкой величине константы скорости восстановления СеW10.
(A) Участок роста тока при Е > -0.3 В. Отметим, что поправка на ток заряжения не очень точна, так как измеряемый ток в начале этого участка сравним с фоновым. Поскольку в данном случае нельзя исключить возможности параллельного окисления ртути, то трудно определить потенциал, при котором ток достигает нулевого значения и однозначно обсуждать его как потенциал начала восстановления исследуемого аниона. В то же время, можно вполне определенно утверждать, что восстановление CeW10 протекает только при потенциалах на ~1 В более отрицательных, чем стандартный потенциал, приведенный в [2,5]. Если данные [5] рассматривать как надежные (Некоторые сомнения вызывает методика титрования [5]: поскольку октациановольфрамат является более прочным комплексом, чем гексацианоферрат, в ходе титрования не исключена деструкция гетерополисоединения с образованием аквакомплексов Се(IV) и, соответственно, смещение плато на титриметрической кривой в сторону более высоких потенциалов.), это различие можно интерпретировать только в предположении о чрезвычайно низкой величине константы скорости восстановления СеW10.
![]() (B) Участок предельного тока. Зависимость предельного тока от высоты ртутного столба h при аппроксимации функцией вида I = a + bhk дает коэффициенты а » 0 и k =0.49±0.02. Пропорциональность тока величине h1/2, а также концентрации реагента в интервале 0.5 - 3 мМ позволяют заключить, что предельный ток является диффузионным. Рассчитанный по уравнению Ильковича в предположении о том, что число переносимых электронов n = 1 (переход Ce(IV/III)) коэффициент диффузии реагента D составляет (6.0 ± 0.5)*10-10 м2/с. Оцененный по уравнению Стокса-Эйнштейна эффективный гидродинамический радиус для исследованного реагента не превышает, соответственно, 0.35 нм, что значимо меньше линейных размеров иона церийвольфрамата вдоль любой из осей (рис.1). Аномально высокие коэффициенты диффузии для гетерополисоединений неоднократно упоминались в литературе [1,12,13] и объяснялись слабой гидратацией и отличием формы ионов от сферической.
(B) Участок предельного тока. Зависимость предельного тока от высоты ртутного столба h при аппроксимации функцией вида I = a + bhk дает коэффициенты а » 0 и k =0.49±0.02. Пропорциональность тока величине h1/2, а также концентрации реагента в интервале 0.5 - 3 мМ позволяют заключить, что предельный ток является диффузионным. Рассчитанный по уравнению Ильковича в предположении о том, что число переносимых электронов n = 1 (переход Ce(IV/III)) коэффициент диффузии реагента D составляет (6.0 ± 0.5)*10-10 м2/с. Оцененный по уравнению Стокса-Эйнштейна эффективный гидродинамический радиус для исследованного реагента не превышает, соответственно, 0.35 нм, что значимо меньше линейных размеров иона церийвольфрамата вдоль любой из осей (рис.1). Аномально высокие коэффициенты диффузии для гетерополисоединений неоднократно упоминались в литературе [1,12,13] и объяснялись слабой гидратацией и отличием формы ионов от сферической.
![]() (C) Участок спада тока. Учитывая анионную природу реагента, можно предположить, что торможение процесса в области отрицательных зарядов ртутного электрода обусловлено возрастающим вкладом отталкивательного взаимодействия реагента с электродом. Это явление широко изучалось ранее [14,15], однако известно лишь ограниченное число систем, для которых удавалось одновременно наблюдать участки роста тока при положительных и снижения тока при отрицательных зарядах поверхности (например, [16]). Независимой причиной ингибирования процесса может быть блокировка поверхности электрода прочно адсорбирующимися на электроде продуктами восстановления. Однако для изучаемой системы последнее объяснение представляется менее вероятным, так как оно не согласуется с существенным увеличением тока в минимуме с ростом концентрации фонового электролита (рис. 2). Это явление типично именно для электростатической природы торможения. При концентрациях фона выше 0,3 М участки (B) и (C) становятся неразличимыми (кривые 4 и 5 рис. 2). К сожалению, проведение измерений при концентрациях фонового электролита ниже 0.01 М не представляется возможным в виду недостаточной буферной емкости ацетатных растворов, поэтому экспериментально не удается проверить возможность еще более выраженного, чем на кривой 1 рис. 2, торможения процесса.
(C) Участок спада тока. Учитывая анионную природу реагента, можно предположить, что торможение процесса в области отрицательных зарядов ртутного электрода обусловлено возрастающим вкладом отталкивательного взаимодействия реагента с электродом. Это явление широко изучалось ранее [14,15], однако известно лишь ограниченное число систем, для которых удавалось одновременно наблюдать участки роста тока при положительных и снижения тока при отрицательных зарядах поверхности (например, [16]). Независимой причиной ингибирования процесса может быть блокировка поверхности электрода прочно адсорбирующимися на электроде продуктами восстановления. Однако для изучаемой системы последнее объяснение представляется менее вероятным, так как оно не согласуется с существенным увеличением тока в минимуме с ростом концентрации фонового электролита (рис. 2). Это явление типично именно для электростатической природы торможения. При концентрациях фона выше 0,3 М участки (B) и (C) становятся неразличимыми (кривые 4 и 5 рис. 2). К сожалению, проведение измерений при концентрациях фонового электролита ниже 0.01 М не представляется возможным в виду недостаточной буферной емкости ацетатных растворов, поэтому экспериментально не удается проверить возможность еще более выраженного, чем на кривой 1 рис. 2, торможения процесса.
![]() (D) Участок резкого роста тока. При потенциалах отрицательнее -1.0 В начинается восстановление оксовольфраматных лигандов и, возможно, каталитическое выделение водорода. Из литературы известно, что начало резкого роста тока выделения водорода на ртути при фиксированном рН сдвигается в сторону более положительных потенциалов при увеличении концентрации гетерополивольфрамата [1]. Это явление наблюдается и в растворах вольфрамата натрия соответствующей концентрации [17].
(D) Участок резкого роста тока. При потенциалах отрицательнее -1.0 В начинается восстановление оксовольфраматных лигандов и, возможно, каталитическое выделение водорода. Из литературы известно, что начало резкого роста тока выделения водорода на ртути при фиксированном рН сдвигается в сторону более положительных потенциалов при увеличении концентрации гетерополивольфрамата [1]. Это явление наблюдается и в растворах вольфрамата натрия соответствующей концентрации [17].
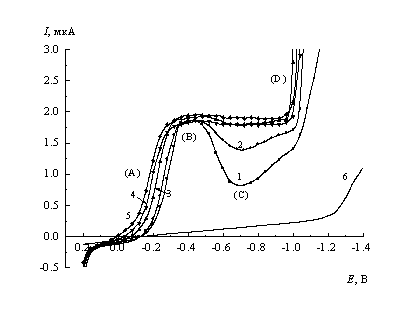 |
| Рис. 2. Полярограммы в растворах 1 мМ Na6H2[CeW10O36] (1-5) с добавками ацетатных буферных смесей (рН = 5.5) с концентрациями, М: 0.01 (1), 0.018 (2), 0.032 (3), 0.056 (4), 0.1 (5), и в растворе 0.1 М фона (6). |
![]() С точки зрения исследований кинетики переноса электрона к центральному иону Ce(IV) наибольший интерес для дальнейшего анализа представляют участки (A) и (C).
С точки зрения исследований кинетики переноса электрона к центральному иону Ce(IV) наибольший интерес для дальнейшего анализа представляют участки (A) и (C).
![]() Следует отметить необычный для восстановления анионов ход кривых восстановления CeW10 на участке (A) (рис. 2): с ростом концентрации фонового электролита при положительных зарядах поверхности ток возрастает, тогда как в рамках теории замедленного разряда следовало бы ожидать противоположного эффекта. Ранее в [18] наблюдался существенный рост тока при увеличении концентрации фона при восстановлении центрального иона Mn(IV) в анионе [MnIVW6O24]8- со структурой Андерсона, однако для экспериментального наблюдения был доступен более узкий интервал потенциалов, непосредственно примыкающий к области растворения ртути.
Следует отметить необычный для восстановления анионов ход кривых восстановления CeW10 на участке (A) (рис. 2): с ростом концентрации фонового электролита при положительных зарядах поверхности ток возрастает, тогда как в рамках теории замедленного разряда следовало бы ожидать противоположного эффекта. Ранее в [18] наблюдался существенный рост тока при увеличении концентрации фона при восстановлении центрального иона Mn(IV) в анионе [MnIVW6O24]8- со структурой Андерсона, однако для экспериментального наблюдения был доступен более узкий интервал потенциалов, непосредственно примыкающий к области растворения ртути.
![]() Можно предположить, что механизмы восстановления аниона CeW10 различны при положительных и отрицательных зарядах поверхности. Согласно [19], в интервале потенциалов 0.0 ¸ -0.5 В различные изополи- и гетерополивольфраматы обнаруживают тенденцию к прочной хемосорбции на ртути, причем имеются свидетельства в пользу того, что образующиеся плотные вольфраматные монослои стабилизируются соадсорбирующимися катионами. Это позволяет обосновать гипотезу о том, что при положительных зарядах поверхности гетерополианионы восстанавливаются из адсорбированного состояния, и скорость восстановления увеличивается с ростом заполнения ими поверхности. Последнее тем выше, чем больше возможность стабилизации анионных слоев катионами, т.е. можно ожидать его роста и, соответственно, ускорения реакции при увеличении концентрации катионов. Данная гипотеза не противоречит интерпретации, предложенной в [18] (участие в процессе ионных ассоциатов реагента с катионами электролита фона в условиях частичного проникновения в плотную часть двойного слоя, т.е. фактически при специфической адсорбции).
Можно предположить, что механизмы восстановления аниона CeW10 различны при положительных и отрицательных зарядах поверхности. Согласно [19], в интервале потенциалов 0.0 ¸ -0.5 В различные изополи- и гетерополивольфраматы обнаруживают тенденцию к прочной хемосорбции на ртути, причем имеются свидетельства в пользу того, что образующиеся плотные вольфраматные монослои стабилизируются соадсорбирующимися катионами. Это позволяет обосновать гипотезу о том, что при положительных зарядах поверхности гетерополианионы восстанавливаются из адсорбированного состояния, и скорость восстановления увеличивается с ростом заполнения ими поверхности. Последнее тем выше, чем больше возможность стабилизации анионных слоев катионами, т.е. можно ожидать его роста и, соответственно, ускорения реакции при увеличении концентрации катионов. Данная гипотеза не противоречит интерпретации, предложенной в [18] (участие в процессе ионных ассоциатов реагента с катионами электролита фона в условиях частичного проникновения в плотную часть двойного слоя, т.е. фактически при специфической адсорбции).
![]() Для более детальной характеристики электрохимического поведения анионов церийдекавольфрамата представляется целесообразным сравнительное исследование растворов, в объеме которых состояние (зарядность) реагента изменяются в достаточно широких пределах, а строение ионного двойного слоя остается неизменным. Поскольку при варьировании концентрации катионов натрия средний заряд в объеме раствора и скачок потенциала в диффузном слое невозможно изменять независимым образом, была предпринята попытка использовать для решения этой задачи смещение равновесия по иону гидроксония в растворах с постоянной ионной силой.
Для более детальной характеристики электрохимического поведения анионов церийдекавольфрамата представляется целесообразным сравнительное исследование растворов, в объеме которых состояние (зарядность) реагента изменяются в достаточно широких пределах, а строение ионного двойного слоя остается неизменным. Поскольку при варьировании концентрации катионов натрия средний заряд в объеме раствора и скачок потенциала в диффузном слое невозможно изменять независимым образом, была предпринята попытка использовать для решения этой задачи смещение равновесия по иону гидроксония в растворах с постоянной ионной силой.
![]() С целью характеристики состояния анионов в растворах CeW10 при различных рН и постоянной ионной силе были зарегистрированы спектры поглощения растворов в диапазоне длин волн 200 - 400 нм. Во всем исследованном интервале рН при концентрации реагента 0.03 - 3 мМ в этом спектральном диапазоне было подтверждено выполнение закона Бугера - Ламберта - Бера.
С целью характеристики состояния анионов в растворах CeW10 при различных рН и постоянной ионной силе были зарегистрированы спектры поглощения растворов в диапазоне длин волн 200 - 400 нм. Во всем исследованном интервале рН при концентрации реагента 0.03 - 3 мМ в этом спектральном диапазоне было подтверждено выполнение закона Бугера - Ламберта - Бера.
![]() На рис. 3 приведены спектры поглощения 0.8 мМ растворов СеW10, содержащих 0.1 М ацетатные буферные смеси. В спектрах можно выделить четыре характерных области с качественно различными рН-зависимостями. С учетом наблюдаемых тенденций можно отнести полосы I и II к двум формам церийдекавольфрамат-иона, различающихся степенью протонирования. Нормированный спектр, приведенный в [8], трудно однозначно сопоставить с данными рис. 3 из-за неопределенности условий проводившихся в [8] измерений. По форме, однако, он находится в хорошем согласии с участком кривой 4 рис.3 при длинах волн 223 - 330 нм (при меньших длинах волн измерения в [8] не проводились). Поскольку в растворах CeW10 без добавок буферных смесей устанавливаются рН 5.8 - 6.2, согласие данных [8] именно со спектром при сравнительно высоких рН в исследованной нами серии представляется разумным.
На рис. 3 приведены спектры поглощения 0.8 мМ растворов СеW10, содержащих 0.1 М ацетатные буферные смеси. В спектрах можно выделить четыре характерных области с качественно различными рН-зависимостями. С учетом наблюдаемых тенденций можно отнести полосы I и II к двум формам церийдекавольфрамат-иона, различающихся степенью протонирования. Нормированный спектр, приведенный в [8], трудно однозначно сопоставить с данными рис. 3 из-за неопределенности условий проводившихся в [8] измерений. По форме, однако, он находится в хорошем согласии с участком кривой 4 рис.3 при длинах волн 223 - 330 нм (при меньших длинах волн измерения в [8] не проводились). Поскольку в растворах CeW10 без добавок буферных смесей устанавливаются рН 5.8 - 6.2, согласие данных [8] именно со спектром при сравнительно высоких рН в исследованной нами серии представляется разумным.
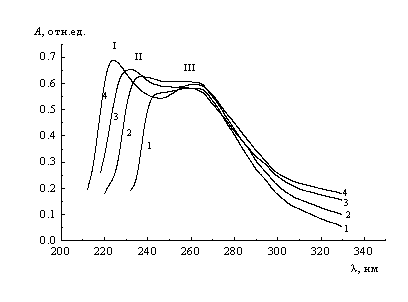 |
| Рис. 3. Спектры поглощения растворов 0.8 мМ Na6H2[CeW10O36] при концентрации ацетатных буферных смесей 0.1 М и рН: 1 - 3.5, 2 - 4.0, 3 - 4.5, 4 - 5.5. |
![]() Полученные нами спектры представляют собой комбинацию как минимум трех гауссовых кривых, и полоса III может отвечать некой форме реагента, содержание которой в растворе в исследованном интервале рН не изменяется. Проследить эволюцию полосы III не удается, поскольку при рН < 3.5 и рН > 6.0 происходит разложение CeW10. Вполне вероятно, что каждая из двух форм, которым отвечают полосы I и II, имеет также вторую полосу поглощения в одной и той же спектральной области III. Ниже мы с целью полуколичественного анализа ограничимся простейшим аддитивным рассмотрением в предположении о присутствии во всех растворах только двух форм аниона с различной степенью протонирования (Вообще говоря, в этом равновесии могут участвовать и ассоциаты СеW10 с катионами натрия. Приведенная оценка рКа дана в предположении о том, что равновесия с участием ионов гидроксония и ионов натрия устанавливаются независимо.):
Полученные нами спектры представляют собой комбинацию как минимум трех гауссовых кривых, и полоса III может отвечать некой форме реагента, содержание которой в растворе в исследованном интервале рН не изменяется. Проследить эволюцию полосы III не удается, поскольку при рН < 3.5 и рН > 6.0 происходит разложение CeW10. Вполне вероятно, что каждая из двух форм, которым отвечают полосы I и II, имеет также вторую полосу поглощения в одной и той же спектральной области III. Ниже мы с целью полуколичественного анализа ограничимся простейшим аддитивным рассмотрением в предположении о присутствии во всех растворах только двух форм аниона с различной степенью протонирования (Вообще говоря, в этом равновесии могут участвовать и ассоциаты СеW10 с катионами натрия. Приведенная оценка рКа дана в предположении о том, что равновесия с участием ионов гидроксония и ионов натрия устанавливаются независимо.):
| H9-z[CeW10O36](z-1)- Û H8-z[CeW10O36]z-+ Н+ | (1) |
![]() Исходя из анализа величин интенсивности в области длин волн I, можно предположить, что при рН = 3.5 и ниже депротонированная форма аниона в растворе практически отсутствует, а при рН > 5.5 ее доля близка к 100%, в то время как протонированная форма практически отсутствует. В рамках этого предположения по серии спектров можно однозначно определить коэффициенты экстинкции двух форм и константу равновесия Ка процесса (1). Расчет приводит к рКа » 4,5.
Исходя из анализа величин интенсивности в области длин волн I, можно предположить, что при рН = 3.5 и ниже депротонированная форма аниона в растворе практически отсутствует, а при рН > 5.5 ее доля близка к 100%, в то время как протонированная форма практически отсутствует. В рамках этого предположения по серии спектров можно однозначно определить коэффициенты экстинкции двух форм и константу равновесия Ка процесса (1). Расчет приводит к рКа » 4,5.
![]() Полярограммы растворов СеW10 демонстрируют практическую независимость хода участков (A) и (B) от рН раствора при постоянной концентрации ацетатной буферной смеси; в то же время на участке (C) наблюдается уменьшение тока с ростом рН (рис.4). Потенциалы начала резкого роста тока на участке (D), соответствующие восстановлению вольфраматных лигандов и ионов гидроксония, чрезвычайно резко зависят от рН. С учетом данных рис. 2 по ускорению процессов в этой области с ростом ионной силы раствора можно уверенно утверждать, что электродные превращения анионных вольфраматных фрагментов вносят в измеренные токи значительный вклад. В то же время, для процессов с переносом 1 -2 электронов трудно предположить последующее присоединение пяти или более протонов, что формально соответствовало бы наблюдаемому смещению волны ~ 0.3 В/рН. Представляется более вероятным, что восстановление W(VI) в CeW10 приводит к деструкции комплекса, в результате которой в исследованном интервале рН образуются различные изополивольфраматы [2]. В этом случае суммарная рН-зависимость может оказаться достаточно сильной, т.к. в нее дополнительно вносят вклад последующие химические стадии.
Полярограммы растворов СеW10 демонстрируют практическую независимость хода участков (A) и (B) от рН раствора при постоянной концентрации ацетатной буферной смеси; в то же время на участке (C) наблюдается уменьшение тока с ростом рН (рис.4). Потенциалы начала резкого роста тока на участке (D), соответствующие восстановлению вольфраматных лигандов и ионов гидроксония, чрезвычайно резко зависят от рН. С учетом данных рис. 2 по ускорению процессов в этой области с ростом ионной силы раствора можно уверенно утверждать, что электродные превращения анионных вольфраматных фрагментов вносят в измеренные токи значительный вклад. В то же время, для процессов с переносом 1 -2 электронов трудно предположить последующее присоединение пяти или более протонов, что формально соответствовало бы наблюдаемому смещению волны ~ 0.3 В/рН. Представляется более вероятным, что восстановление W(VI) в CeW10 приводит к деструкции комплекса, в результате которой в исследованном интервале рН образуются различные изополивольфраматы [2]. В этом случае суммарная рН-зависимость может оказаться достаточно сильной, т.к. в нее дополнительно вносят вклад последующие химические стадии.
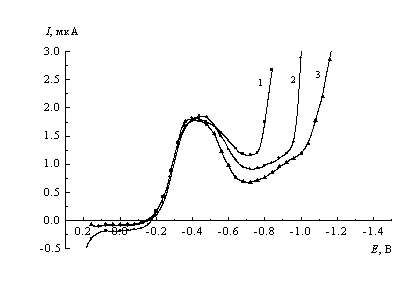 |
| Рис. 4. Полярограммы восстановления в 0.01 М ацетатном буфере в присутствии 1 мМ Na6H2[CeW10O36] при рН: 1 - 4.5, 2 - 5.0, 3 - 5.5. |
![]() Полученные данные для участка (C) находятся в согласии с предположением о параллельном восстановлении нескольких форм реагента разной зарядности в исследуемых растворах. В рамках этого предположения и представлений теории замедленного разряда отталкивание анионов от отрицательно заряженного электрода тем сильнее, чем больше по абсолютной величине их заряд. При смещении равновесия (1) вправо (с ростом рН) доля реагента с более отрицательным зарядом в объеме раствора увеличивается, так же как и соответствующий вклад в суммарный ток. Одновременно вклад реагента с менее отрицательным зарядом, испытывающего менее сильное отталкивание, уменьшается, и, соответственно, суммарный ток падает. Качественно эта трактовка остается справедливой и при сосуществовании в объеме раствора трех и более форм реагента. Необходимо отметить, что ионная сила буферных растворов одной и той же концентрации с ростом рН увеличивается, и при постоянстве заряда реагента это должно было бы приводить к увеличению тока в минимуме с ростом рН. Таким образом, эффект изменения заряда реагента явно преобладает над эффектом противоположного знака, связанного с изменением буферной силы растворов в рассматриваемой серии.
Полученные данные для участка (C) находятся в согласии с предположением о параллельном восстановлении нескольких форм реагента разной зарядности в исследуемых растворах. В рамках этого предположения и представлений теории замедленного разряда отталкивание анионов от отрицательно заряженного электрода тем сильнее, чем больше по абсолютной величине их заряд. При смещении равновесия (1) вправо (с ростом рН) доля реагента с более отрицательным зарядом в объеме раствора увеличивается, так же как и соответствующий вклад в суммарный ток. Одновременно вклад реагента с менее отрицательным зарядом, испытывающего менее сильное отталкивание, уменьшается, и, соответственно, суммарный ток падает. Качественно эта трактовка остается справедливой и при сосуществовании в объеме раствора трех и более форм реагента. Необходимо отметить, что ионная сила буферных растворов одной и той же концентрации с ростом рН увеличивается, и при постоянстве заряда реагента это должно было бы приводить к увеличению тока в минимуме с ростом рН. Таким образом, эффект изменения заряда реагента явно преобладает над эффектом противоположного знака, связанного с изменением буферной силы растворов в рассматриваемой серии.
![]() Для проверки разумности гипотезы о параллельном восстановлении только двух форм реагента была выполнена следующая приближенная оценка. В экспериментальные значения тока I была внесена формальная поправка на концентрационную поляризацию:
Для проверки разумности гипотезы о параллельном восстановлении только двух форм реагента была выполнена следующая приближенная оценка. В экспериментальные значения тока I была внесена формальная поправка на концентрационную поляризацию:
| Iet = IId/(Id - I) | (2) |
где Iet - суммарный ток переноса электрона. Представляя эту величину как сумму токов восстановления депротонированной (I1) (4а) и протонированной (I2) (4б) форм, можно записать следующие соотношения:
| Iet = I1 + I2, | (3) |
| I1 = const1c1exp((a+z)Fy1/RT) | (4а) |
| I2 = const2 c2 exp((a+(z-1))Fy1/RT), | (4b) |
где с1 и с2 - объемные концентрации обсуждаемых форм с зарядами z- и (z-1)-, соответственно, y1 - эффективное значение пси-прим-потенциала, a - коэффициент переноса. Поскольку существенное влияние протонирования оксоокружения центрального иона на величины трансмиссионного коэффициента и полной энергии реорганизации маловероятно, можно предположить, что константы скорости для обеих обсуждаемых форм близки. Поэтому из (3), (4а) и (4б), полагая const1 = const2 = const, можно получить для суммарного тока, вне зависимости от предположения о величине z, следующее соотношение:
| Iet = const exp((a+z)Fy1/RT)(c1+c2 exp(-Fy1/RT)). | (5) |
![]() Допустим, что восстановление обеих форм протекает без проникновения в плотную часть двойного слоя, и рассчитаем эффективный пси-прим-потенциал, при котором уравнение (5) приводит к найденному экспериментально соотношению токов в минимуме (при Е = -0.6 В): Iet (pH 5,5): Iet (pH 5,0): Iet (pH 4,5)=1:1.81:3.07.
Допустим, что восстановление обеих форм протекает без проникновения в плотную часть двойного слоя, и рассчитаем эффективный пси-прим-потенциал, при котором уравнение (5) приводит к найденному экспериментально соотношению токов в минимуме (при Е = -0.6 В): Iet (pH 5,5): Iet (pH 5,0): Iet (pH 4,5)=1:1.81:3.07.
![]() Подставляя a = 0,5 и (для каждого значения рН) соотношения с1 и с2, рассчитанные из полученной из спектров поглощения величины Ka, получаем, что такое соотношение токов соответствует y1 = -0.06 В. Концентрации катионов фона (если пренебречь при столь грубой оценке эффектами изменения ионной силы) составляет 0.016 М. Интерполяция данных [20] для Е = -0.6 В (с учетом поправки на шкалу потенциала) дает для указанной концентрации потенциал внешней плоскости Гельмгольца, всего на несколько мВ превышающий найденное значение y1.
Подставляя a = 0,5 и (для каждого значения рН) соотношения с1 и с2, рассчитанные из полученной из спектров поглощения величины Ka, получаем, что такое соотношение токов соответствует y1 = -0.06 В. Концентрации катионов фона (если пренебречь при столь грубой оценке эффектами изменения ионной силы) составляет 0.016 М. Интерполяция данных [20] для Е = -0.6 В (с учетом поправки на шкалу потенциала) дает для указанной концентрации потенциал внешней плоскости Гельмгольца, всего на несколько мВ превышающий найденное значение y1.
![]() Таким образом, предположения о существовании в исследуемых растворах двух форм реагента, различающихся по степени протонирования, и об их параллельном внешнесферном восстановлении при отрицательных зарядах поверхности ртути, по крайней мере, не противоречат наблюдаемым фактам. Разумеется, проведенные выше оценки нельзя (как и целый ряд других, выполненных в рамках теории замедленного разряда) рассматривать как однозначные. Существенная неопределенность связана с соотношением модельных параметров (коэффициента переноса и пси-прим-потенциала), а дополнительная неопределенность в величине последнего для какой-либо локализации обусловлена большими размерами и сложными зарядовыми распределениями частиц реагентов [21]. Кроме того, открытым остается вопрос о рН-зависимости равновесного потенциала системы Ce(IV/III)W10. Если такая зависимость существует, в оценках следует учитывать также различия перенапряжения для форм реагента с различными степенями протонирования. Наконец, поправка на концентрационную поляризацию по уравнению (2) в условиях параллельного восстановления нескольких реагентов разной зарядности может приводить к существенному занижению Iet.
Таким образом, предположения о существовании в исследуемых растворах двух форм реагента, различающихся по степени протонирования, и об их параллельном внешнесферном восстановлении при отрицательных зарядах поверхности ртути, по крайней мере, не противоречат наблюдаемым фактам. Разумеется, проведенные выше оценки нельзя (как и целый ряд других, выполненных в рамках теории замедленного разряда) рассматривать как однозначные. Существенная неопределенность связана с соотношением модельных параметров (коэффициента переноса и пси-прим-потенциала), а дополнительная неопределенность в величине последнего для какой-либо локализации обусловлена большими размерами и сложными зарядовыми распределениями частиц реагентов [21]. Кроме того, открытым остается вопрос о рН-зависимости равновесного потенциала системы Ce(IV/III)W10. Если такая зависимость существует, в оценках следует учитывать также различия перенапряжения для форм реагента с различными степенями протонирования. Наконец, поправка на концентрационную поляризацию по уравнению (2) в условиях параллельного восстановления нескольких реагентов разной зарядности может приводить к существенному занижению Iet.
![]() Отсутствие влияния рН на ход участков полярограмм при положительных зарядах поверхности ртути не может рассматриваться как свидетельство независимости от рН равновесного потенциала, лежащего вне области идеальной поляризуемости ртутного электрода. В этой связи важной представляется попытка оценки этой величины и ее зависимости от рН на твердых электродах.
Отсутствие влияния рН на ход участков полярограмм при положительных зарядах поверхности ртути не может рассматриваться как свидетельство независимости от рН равновесного потенциала, лежащего вне области идеальной поляризуемости ртутного электрода. В этой связи важной представляется попытка оценки этой величины и ее зависимости от рН на твердых электродах.
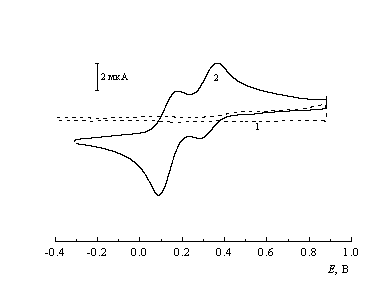
![]() Вольтамперограммы исследуемого реагента на электродах из пирографита обнаруживают существенную зависимость как от рН раствора, так и от природы поверхности материала. Наиболее четкие пики тока в интервале Е = 0.0 - 0.4 В удается зарегистрировать на базисной грани высокоориентированного пирографита (HOPG) с плотностью моноатомных ступеней порядка 1 мкм-1 (линейные размеры террас заведомо гораздо больше, чем размер частиц реагента). Воспроизводимость откликов от скола к сколу не является вполне количественной. Как правило (особенно при рН 5.5 - 6.0), наблюдаются две пары асимметричных пиков, причем для обеих пар разность потенциалов анодного и катодного пиков не превышает 100 мВ (см. пример на рис. 5), в некоторых случаях надежно фиксируется лишь одна пара пиков. Величины формальных потенциалов (Эти величины определяли как средние между величинами потенциалов анодного и катодного пиков. В интервале скоростей развертки 10 - 100 мВ/с формальные потенциалы оставались постоянными с точностью до 5 мВ.) зависят от рН и составляют при рН = 5.5 около 0.15 и около 0.3 В, т.е. отвечают гораздо более положительным значениям, чем возможные потенциалы восстановления вольфраматных лигандов. Поэтому отнесение регистрируемых пиков к переходам Се(IV/III) представляется вполне надежным.
Вольтамперограммы исследуемого реагента на электродах из пирографита обнаруживают существенную зависимость как от рН раствора, так и от природы поверхности материала. Наиболее четкие пики тока в интервале Е = 0.0 - 0.4 В удается зарегистрировать на базисной грани высокоориентированного пирографита (HOPG) с плотностью моноатомных ступеней порядка 1 мкм-1 (линейные размеры террас заведомо гораздо больше, чем размер частиц реагента). Воспроизводимость откликов от скола к сколу не является вполне количественной. Как правило (особенно при рН 5.5 - 6.0), наблюдаются две пары асимметричных пиков, причем для обеих пар разность потенциалов анодного и катодного пиков не превышает 100 мВ (см. пример на рис. 5), в некоторых случаях надежно фиксируется лишь одна пара пиков. Величины формальных потенциалов (Эти величины определяли как средние между величинами потенциалов анодного и катодного пиков. В интервале скоростей развертки 10 - 100 мВ/с формальные потенциалы оставались постоянными с точностью до 5 мВ.) зависят от рН и составляют при рН = 5.5 около 0.15 и около 0.3 В, т.е. отвечают гораздо более положительным значениям, чем возможные потенциалы восстановления вольфраматных лигандов. Поэтому отнесение регистрируемых пиков к переходам Се(IV/III) представляется вполне надежным.
 |
| Рис. 5. Вольтамперограммы в растворе 3 мМ Na6Н2[CeW10O36] + 0.1 M (CH3COOH + CH3COONa) на высокоориентированном графите при рН 5.5. |
![]() Не вызывает сомнений факт прочной хемосорбции церийдекавольфрамата на графите. Это демонстрируют опыты с отмывкой электрода из HOPG фоновым буферным раствором после выдержки в растворе CeW10 при разомкнутой цепи или кратковременного циклирования в нем. На отмытом электроде в растворе фона регистрируются выраженные, хотя и меньшие по величине пики тока в той же области потенциалов. Им соответствуют заряды около 5 мкКл/cм2, что отвечает обратимому редокс-превращению иммобилизованного Се(IV/III) в субмонослойных количествах ( Если число гетерополианионов в плотном монослое определять по кристаллохимическим данным [3], уподобив адсорбат двумерной решетке, то оценка заряда, отвечающего переходу Се(IV/III) в монослое, дает 20 - 25 мкКл/см2.).
Не вызывает сомнений факт прочной хемосорбции церийдекавольфрамата на графите. Это демонстрируют опыты с отмывкой электрода из HOPG фоновым буферным раствором после выдержки в растворе CeW10 при разомкнутой цепи или кратковременного циклирования в нем. На отмытом электроде в растворе фона регистрируются выраженные, хотя и меньшие по величине пики тока в той же области потенциалов. Им соответствуют заряды около 5 мкКл/cм2, что отвечает обратимому редокс-превращению иммобилизованного Се(IV/III) в субмонослойных количествах ( Если число гетерополианионов в плотном монослое определять по кристаллохимическим данным [3], уподобив адсорбат двумерной решетке, то оценка заряда, отвечающего переходу Се(IV/III) в монослое, дает 20 - 25 мкКл/см2.).
![]() По-видимому, редокс-переходы в исследуемой системе протекают с участием адсорбированных интермедиатов, и не слишком высокая количественная воспроизводимость откликов связана с высокой чувствительностью адсорбционного поведения СеW10 к состоянию поверхности HOPG, несколько изменяющемуся от скола к сколу. Учитывая приведенные выше спектральные данные и оценки рКа, раздвоение пиков при сравнительно высоких рН вряд ли можно отнести к восстановлению двух различных форм реагента. В то же время нельзя исключить, что процесс осложнен стадией протонирования восстановленной формы, и стадийные механизмы прямого и обратного процессов различаются - при некоторых соотношениях скоростей стадий протонирования и переноса электрона в этом случае также можно ожидать вольтамперограмм сложной формы.
По-видимому, редокс-переходы в исследуемой системе протекают с участием адсорбированных интермедиатов, и не слишком высокая количественная воспроизводимость откликов связана с высокой чувствительностью адсорбционного поведения СеW10 к состоянию поверхности HOPG, несколько изменяющемуся от скола к сколу. Учитывая приведенные выше спектральные данные и оценки рКа, раздвоение пиков при сравнительно высоких рН вряд ли можно отнести к восстановлению двух различных форм реагента. В то же время нельзя исключить, что процесс осложнен стадией протонирования восстановленной формы, и стадийные механизмы прямого и обратного процессов различаются - при некоторых соотношениях скоростей стадий протонирования и переноса электрона в этом случае также можно ожидать вольтамперограмм сложной формы.
![]() Отметим, что, несмотря на недостаточную точность определения формальных потенциалов по вольтамперограммам на прижимном донном электроде из HOPG, однозначным является вывод об их существенно меньших значениях по сравнению с приведенным в [2,5]. Поэтому можно уверенно утверждать, что потенциал начала наблюдаемого восстановления CeW10 на ртутном электроде смещен относительно равновесного потенциала не более чем на 0.3-0.5 В, т.е. кривые на рис. 2 и 4 охватывают область перенапряжений не более чем до 1.0 В. Это существенно отличает исследуемый процесс от широко известных модельных реакций восстановления анионов при высоких перенапряжениях [14,15].
Отметим, что, несмотря на недостаточную точность определения формальных потенциалов по вольтамперограммам на прижимном донном электроде из HOPG, однозначным является вывод об их существенно меньших значениях по сравнению с приведенным в [2,5]. Поэтому можно уверенно утверждать, что потенциал начала наблюдаемого восстановления CeW10 на ртутном электроде смещен относительно равновесного потенциала не более чем на 0.3-0.5 В, т.е. кривые на рис. 2 и 4 охватывают область перенапряжений не более чем до 1.0 В. Это существенно отличает исследуемый процесс от широко известных модельных реакций восстановления анионов при высоких перенапряжениях [14,15].
![]() В отсутствие осложнений, связанных с прижимной конструкцией электрода, измерения удалось выполнить лишь на частично разупорядоченном ориентированном пирографите, краевые фрагменты которого характеризуются более высокой плотностью ступеней. Этот электрод имеет значительно большую общую поляризационную емкость, чем HOPG, и на нем удается надежно наблюдать только один рН-зависимый редокс-переход. Отвечающие ему формальные потенциалы при заданных рН не отличаются существенно от аналогичных величин, определенных на HOPG, занимая промежуточное положение между двумя упомянутыми выше формальными потенциалами (0.25 В при рН = 5.5). Формально обратимость редокс-переходов при рН 5.5 - 6.0 оказывается достаточно высокой, т.к. разность потенциалов пиков близка к 0.06 В, но во всем исследованном интервале потенциалов емкость электрода в растворах CeW10 остается в 2-4 раза выше фоновой (тогда как на HOPG вне области потенциалов пиков емкость отвечает фоновым значениям, рис. 5). Поскольку как высоты пиков, так и "избыточная емкость" увеличиваются с концентрацией CeW10, это свидетельствует о том, что превращения исследуемого реагента на разупорядоченном пирографите происходят в достаточно широкой области потенциалов, например, восстанавливаются и окисляются множество адсорбированных форм реагента с различными энергиями связи, а формальный потенциал при этом чувствителен к трансформации частиц при адсорбции. Нельзя также исключить взаимодействия адсорбата(ов) CeW10 с функциональными группами на углеродной поверхности.
В отсутствие осложнений, связанных с прижимной конструкцией электрода, измерения удалось выполнить лишь на частично разупорядоченном ориентированном пирографите, краевые фрагменты которого характеризуются более высокой плотностью ступеней. Этот электрод имеет значительно большую общую поляризационную емкость, чем HOPG, и на нем удается надежно наблюдать только один рН-зависимый редокс-переход. Отвечающие ему формальные потенциалы при заданных рН не отличаются существенно от аналогичных величин, определенных на HOPG, занимая промежуточное положение между двумя упомянутыми выше формальными потенциалами (0.25 В при рН = 5.5). Формально обратимость редокс-переходов при рН 5.5 - 6.0 оказывается достаточно высокой, т.к. разность потенциалов пиков близка к 0.06 В, но во всем исследованном интервале потенциалов емкость электрода в растворах CeW10 остается в 2-4 раза выше фоновой (тогда как на HOPG вне области потенциалов пиков емкость отвечает фоновым значениям, рис. 5). Поскольку как высоты пиков, так и "избыточная емкость" увеличиваются с концентрацией CeW10, это свидетельствует о том, что превращения исследуемого реагента на разупорядоченном пирографите происходят в достаточно широкой области потенциалов, например, восстанавливаются и окисляются множество адсорбированных форм реагента с различными энергиями связи, а формальный потенциал при этом чувствителен к трансформации частиц при адсорбции. Нельзя также исключить взаимодействия адсорбата(ов) CeW10 с функциональными группами на углеродной поверхности.
![]() Снижение рН приводит к резкому усилению необратимости редокс-перехода и смещению как анодного, так и катодного пиков в сторону более положительных значений, так, что при рН около 4 пики тока наблюдать не удается (из-за наложения анодного фарадеевского процесса на подложке). В узкой области рН, доступной для регистрации пиков тока, формальные потенциалы смещаются на 0.15 - 0.18 В/рН. Однако значение рН = 6.0 близко к границе области устойчивочти комплекса, щелочной гидролиз которого протекает с заметной скоростью при рН > 7, поэтому нельзя исключить, что истинная зависимость равновесного потенциала от рН является более слабой.
Снижение рН приводит к резкому усилению необратимости редокс-перехода и смещению как анодного, так и катодного пиков в сторону более положительных значений, так, что при рН около 4 пики тока наблюдать не удается (из-за наложения анодного фарадеевского процесса на подложке). В узкой области рН, доступной для регистрации пиков тока, формальные потенциалы смещаются на 0.15 - 0.18 В/рН. Однако значение рН = 6.0 близко к границе области устойчивочти комплекса, щелочной гидролиз которого протекает с заметной скоростью при рН > 7, поэтому нельзя исключить, что истинная зависимость равновесного потенциала от рН является более слабой.
![]() Отмеченная на качественном уровне корреляция степени "размытости" пиков на вольтамперограммах и качества поверхности графита может быть интерпретирована в предположении о преимущественной сильной хемосорбции CeW10 на ступенях и слабой адсорбируемости (или ее отсутствии) на террасах. По-видимому, существуют фрагменты поверхности, на которых адсорбция не слишком сильна, и редокс-переход с участием адсорбата происходит достаточно быстро. В этом случае две пары хорошо выраженных на HOPG пиков, которые сохраняются и после отмывки электрода, могут относиться, например, к электродным процессам с участием адсорбатов на ступенях и террасах или частиц, адсорбированных в разных ориентациях.
Отмеченная на качественном уровне корреляция степени "размытости" пиков на вольтамперограммах и качества поверхности графита может быть интерпретирована в предположении о преимущественной сильной хемосорбции CeW10 на ступенях и слабой адсорбируемости (или ее отсутствии) на террасах. По-видимому, существуют фрагменты поверхности, на которых адсорбция не слишком сильна, и редокс-переход с участием адсорбата происходит достаточно быстро. В этом случае две пары хорошо выраженных на HOPG пиков, которые сохраняются и после отмывки электрода, могут относиться, например, к электродным процессам с участием адсорбатов на ступенях и террасах или частиц, адсорбированных в разных ориентациях.
![]() Электрохимические превращения CeW10 на поликристаллическом золотом электроде оказываются в еще большей степени осложнены адсорбционными явлениями. Типичная вольтамперограмма для золотого электрода приведена на рис. 6а (рН 6,0). При рН 6.0 и потенциалах отрицательнее 0.2 В фиксируется волна восстановления комплекса, а на обратном ходе - размытый пик анодного тока при Е » 0.4 В (кривые 2 - 4 рис. 6а). На обоих указанных участках ток пропорционален объемной концентрации реагента (рис. 6б) и квадратному корню из скорости развертки (рис. 6в), что позволяет отнести наблюдаемый необратимый отклик к исследуемому процессу. Анодная ветвь вольтамперограммы отвечает области положительных свободных зарядов золота. В пользу прочной хемосорбции частиц СeW10 в этой области свидетельствует существенное подавление адсорбции кислорода на золоте по сравнению с адсорбцией кислорода в растворе фона (кривая 1 рис. 6а). Как показано в [23], уменьшение заряда в кислородной области вдвое может отвечать предельному заполнению поверхности гетерополианионами (предполагается, что образуемая последними двумерная решетка является достаточно рыхлой, и кислород может соадсорбироваться с плотноупакованными поливольфраматами). Слабая зависимость степени подавления адсорбции кислорода от концентрации СеW10 в растворе также свидетельствует в пользу достижения предельного заполнения.
Электрохимические превращения CeW10 на поликристаллическом золотом электроде оказываются в еще большей степени осложнены адсорбционными явлениями. Типичная вольтамперограмма для золотого электрода приведена на рис. 6а (рН 6,0). При рН 6.0 и потенциалах отрицательнее 0.2 В фиксируется волна восстановления комплекса, а на обратном ходе - размытый пик анодного тока при Е » 0.4 В (кривые 2 - 4 рис. 6а). На обоих указанных участках ток пропорционален объемной концентрации реагента (рис. 6б) и квадратному корню из скорости развертки (рис. 6в), что позволяет отнести наблюдаемый необратимый отклик к исследуемому процессу. Анодная ветвь вольтамперограммы отвечает области положительных свободных зарядов золота. В пользу прочной хемосорбции частиц СeW10 в этой области свидетельствует существенное подавление адсорбции кислорода на золоте по сравнению с адсорбцией кислорода в растворе фона (кривая 1 рис. 6а). Как показано в [23], уменьшение заряда в кислородной области вдвое может отвечать предельному заполнению поверхности гетерополианионами (предполагается, что образуемая последними двумерная решетка является достаточно рыхлой, и кислород может соадсорбироваться с плотноупакованными поливольфраматами). Слабая зависимость степени подавления адсорбции кислорода от концентрации СеW10 в растворе также свидетельствует в пользу достижения предельного заполнения.
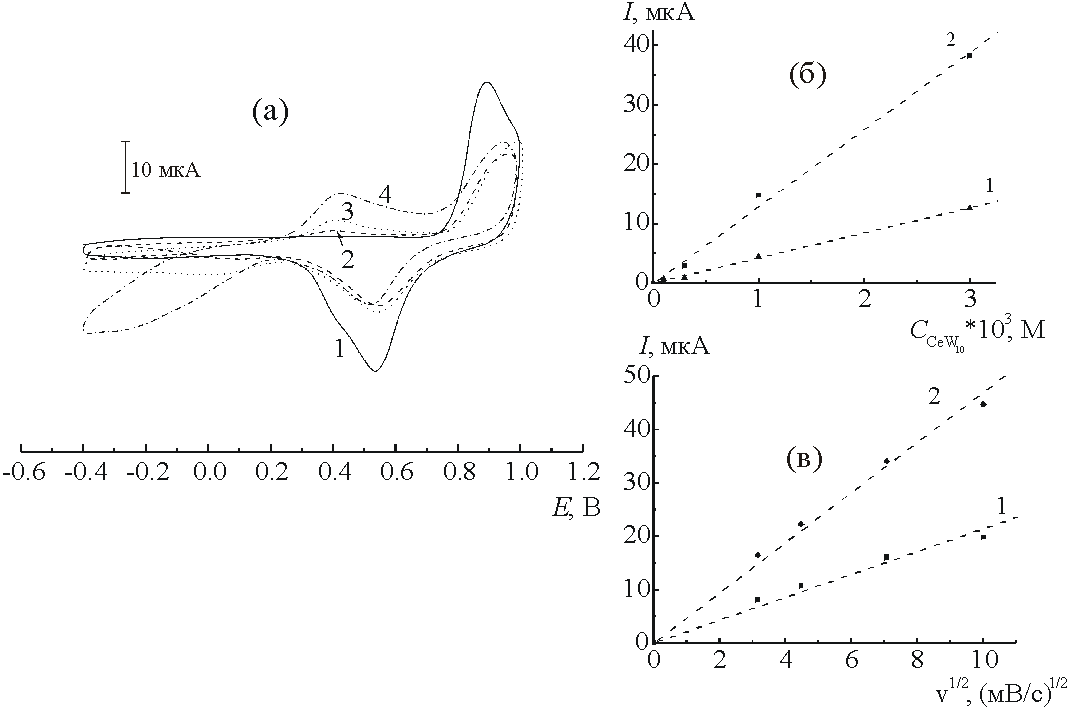 |
| Рис. 6. (a) - Вольтамперограммы, регистрируемые на поликристаллическом золотом электроде в растворе 0.1 M (CH3COOH + CH3COONa) с добавками x мМ Na6Н2[CeW10O36] при рН = 6.0 и скорости развертки 0.05 В/с, x: (1) - 0, (2) - 0.1, (3) - 0.3, (4) - 1.
(б) - Зависимость величины токов при потенциалах анодной (Е = 0.70 В) (1) и катодной (Е = -0.38 В) (2) волн от концентрации Na6H2[CeW10O3]. (в) - Зависимость величины токов в максимуме анодного (Е около 0.40 В) (1) и катодного (Е около -0.35 В) (2) пиков, от v1/2, измеренные для добавки 3 мМ Na6Н2[CeW10O36]. |
![]() Заключение о прочной хемосорбции СеW10 на золоте при Е > 0.2 ¸ 0.3 В хорошо согласуется с предварительными данными ex situ и in situ туннельной микроскопии для поликристаллического золота и текстурированных пленок Au(111). Согласно этим данным, аномально высокая подвижность поверхностных атомов золота в присутствии CeW10 сохраняется и при потенциалах начального участка катодной волны (до -0.2 В), что косвенно указывает на неполную десорбцию реагента даже при отрицательных свободных зарядах поверхности.
Заключение о прочной хемосорбции СеW10 на золоте при Е > 0.2 ¸ 0.3 В хорошо согласуется с предварительными данными ex situ и in situ туннельной микроскопии для поликристаллического золота и текстурированных пленок Au(111). Согласно этим данным, аномально высокая подвижность поверхностных атомов золота в присутствии CeW10 сохраняется и при потенциалах начального участка катодной волны (до -0.2 В), что косвенно указывает на неполную десорбцию реагента даже при отрицательных свободных зарядах поверхности.
![]() Особенностью восстановления CeW10 на золотом электроде является слабая зависимость вольтамперометрических откликов от рН раствора. При рН < 5.5 пики смещаются не более чем на 15-20 мВ/рН, причем их потенциалы немного сближаются. При рН 5.5 - 6.0 качественно эффект остается таким же (обратимость процесса несколько увеличивается), смещения пиков несколько сильнее, но в целом, эффект рН остается слабее, чем на пирографите. Это можно рассматривать как свидетельство принципиальных отличий реакционных слоев на углеродной и золотой поверхностях. По-видимому, при прочной адсорбции протонодонорные свойства реагента существенно изменяются, а смещение формального потенциала при изменении рН достаточно мало по сравнению с очень большой разностью потенциалов анодного и катодного пиков для столь необратимого процесса. В этом смысле прослеживается аналогия в кинетике восстановления Ce(IV)W10 на ртути при положительных зарядах поверхности и на золоте.
Особенностью восстановления CeW10 на золотом электроде является слабая зависимость вольтамперометрических откликов от рН раствора. При рН < 5.5 пики смещаются не более чем на 15-20 мВ/рН, причем их потенциалы немного сближаются. При рН 5.5 - 6.0 качественно эффект остается таким же (обратимость процесса несколько увеличивается), смещения пиков несколько сильнее, но в целом, эффект рН остается слабее, чем на пирографите. Это можно рассматривать как свидетельство принципиальных отличий реакционных слоев на углеродной и золотой поверхностях. По-видимому, при прочной адсорбции протонодонорные свойства реагента существенно изменяются, а смещение формального потенциала при изменении рН достаточно мало по сравнению с очень большой разностью потенциалов анодного и катодного пиков для столь необратимого процесса. В этом смысле прослеживается аналогия в кинетике восстановления Ce(IV)W10 на ртути при положительных зарядах поверхности и на золоте.
![]() Одной из гипотез, объясняющих аномалии восстановления на золотом электроде, является предположение о химическом взаимодействии CeW10 с золотом (выраженный перенос заряда) и образование в поверхностном слое солеподобного соединения, включающего катионы золота и анионы Сe(III)W10. В этом случае анодный процесс мог бы представлять собой окисление Ce(III) с разрушением такой соли, а катодный процесс с участием менее прочно связанных анионов в диффузной части двойного слоя оказался бы существенно заторможен из-за блокировки поверхности солеподобным слоем.
Одной из гипотез, объясняющих аномалии восстановления на золотом электроде, является предположение о химическом взаимодействии CeW10 с золотом (выраженный перенос заряда) и образование в поверхностном слое солеподобного соединения, включающего катионы золота и анионы Сe(III)W10. В этом случае анодный процесс мог бы представлять собой окисление Ce(III) с разрушением такой соли, а катодный процесс с участием менее прочно связанных анионов в диффузной части двойного слоя оказался бы существенно заторможен из-за блокировки поверхности солеподобным слоем.
![]() В настоящей работе дана общая характеристика электрохимического поведения реагента, для которого процесс одноэлектронного восстановления является достаточно медленным в широком интервале потенциалов и свободных зарядов поверхности. На наш взгляд, наибольший интерес представляет специфика восстановления в условиях прочной адсорбции на положительно заряженной поверхности.
В настоящей работе дана общая характеристика электрохимического поведения реагента, для которого процесс одноэлектронного восстановления является достаточно медленным в широком интервале потенциалов и свободных зарядов поверхности. На наш взгляд, наибольший интерес представляет специфика восстановления в условиях прочной адсорбции на положительно заряженной поверхности.
![]() Есть основания полагать, что многие кислород-содержащие анионы и комплексные анионы с акцепторными лигандами при положительных зарядах поверхности склонны к хемосорбции, и их электродные превращения носят поэтому внутрисферный характер (см. например, [24-26]). В этом плане комплекс CeW10, хемосорбция которого выражена чрезвычайно сильно, может представлять интерес как модельный объект, в первую очередь, для исследования роли соадсорбции с фоновыми катионами (адсорбции ассоциатов) в наблюдаемой при положительных зарядах электрода аномальной зависимости тока восстановления от концентрации электролита фона.
Есть основания полагать, что многие кислород-содержащие анионы и комплексные анионы с акцепторными лигандами при положительных зарядах поверхности склонны к хемосорбции, и их электродные превращения носят поэтому внутрисферный характер (см. например, [24-26]). В этом плане комплекс CeW10, хемосорбция которого выражена чрезвычайно сильно, может представлять интерес как модельный объект, в первую очередь, для исследования роли соадсорбции с фоновыми катионами (адсорбции ассоциатов) в наблюдаемой при положительных зарядах электрода аномальной зависимости тока восстановления от концентрации электролита фона.
![]() Работа выполнена при поддержке РФФИ, проект № 02-03-33321а, и частичной поддержке ИНТАС, проект № 99-1093.
Работа выполнена при поддержке РФФИ, проект № 02-03-33321а, и частичной поддержке ИНТАС, проект № 99-1093.